
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Методика электрохимического окисления иодид-ионов при градуировке и анализе ⇐ ПредыдущаяСтр 2 из 2
Аликвоту рабочего раствора А помещают в электрохимическую ячейку. В катод заливают фиксированное количество катодного раствора. В ходе электролиза над раствором продувают газообразный азот. На потенциостате выставляют режим “Потенциал” и ведут электролиз данного раствора в течение определённого времени при фиксированном потенциале, постоянно перемешивая раствор. После окончания электролиза снимаем показания с интегратора кулонометрического.
3.4 Методика электрохимического окисления бромид-ионов при градуировке и анализе
Аликвоту рабочего раствора В помещают в электрохимическую ячейку, в катод заливают фиксированное количество катодного раствора. В ходе электролиза над раствором продувают газообразный азот. На потенциостате выставляют режим “Потенциал” и ведут электролиз данного раствора в течение определённого времени при потенциале выделения йода, постоянно перемешивая раствор. Затем прекращают электролиз, раствор из ячейки переносят в делительную воронку. Извлекают образовавшийся иод двумя последовательными экстракциями по 2 мин с фиксированным количеством CCl4. Органическую фазу удаляют, из водной фазы отбирают аликвоту раствора и помещают в мерную колбу ёмкостью 100 мл, добавляют аликвоту раствора хлорной кислоты и доливают до метки бидистиллированой водой, насыщенной газообразным азотом. В катод заливают фиксированное количество катодного раствора. Аликвоту приготовленного раствора помещают в ячейку, на потенциостате выставляют режим “Потенциал” и ведут электролиз данного раствора в течение определённого времени при фиксированном потенциале, постоянно перемешивая раствор. В ходе электролиза над раствором продувают газообразный азот. После окончания электролиза снимаем показания с интегратора кулонометрического.
Методика электрохимического окисления смеси бромид- и иодид - ионов при градуировке и анализе
Аликвоту рабочего раствора С помещают в электрохимическую ячейку. В катод заливают фиксированное количество катодного раствора. В ходе электролиза над раствором продувают газообразный азот. На потенциостате выставляют режим “Потенциал” и ведут электролиз данного раствора в течение определённого времени при потенциале выделения иода, постоянно перемешивая раствор. Затем прекращают электролиз снимают показания с интегратора кулонометрического. Раствор из ячейки переносят в делительную воронку. Извлекают образовавшийся иод двумя последовательными экстракциями по 2 мин с фиксированным количеством CCl4. Органическую фазу удаляют, из водной фазы отбирают аликвоту раствора и помещают в мерную колбу ёмкостью 100 мл, добавляют аликвоту раствора хлорной кислоты и доливают до метки бидистиллированой водой, насыщенной газообразным азотом. В катод заливают фиксированное количество катодного раствора. Аликвоту приготовленного раствора помещают в ячейку, на потенциостате выставляют режим “Потенциал” и ведут электролиз данного раствора в течение определённого времени при фиксированном потенциале, постоянно перемешивая раствор. В ходе электролиза над раствором продувают газообразный азот. После окончания электролиза снимаем показания с интегратора кулонометрического.
Методика электрохимического окисления смеси бромид- и иодид - ионов при градуировке и анализе без отделения йода
Аликвоту рабочего раствора Д помещают в электрохимическую ячейку. В катод заливают фиксированное количество катодного раствора. В ходе электролиза над раствором продувают газообразный азот. На потенциостате выставляют режим “Потенциал” и ведут электролиз данного раствора в течение 15 мин при потенциале выделения йода (U=+0.75V), постоянно перемешивая раствор. Затем прекращают электролиз, снимают показания с интегратора кулонометрического. Чистят поверхность электродов. Другую аликвоту рабочего раствора Д помещают в электрохимическую ячейку. В катод заливают фиксированное количество катодного раствора. В ходе электролиза над раствором продувают газообразный азот. Электролиз данной пробы ведут в течение 1 часа при потенциале выделения брома (U=+1.1V), постоянно перемешивая раствор. В процессе электролиза снимают показания с интегратора кулонометрического через определенные промежутки времени. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Метод потенциостатической кулонометрии относится к группе электрохимических методов и является абсолютным. Он позволяет судить о содержании анализируемого вещества в растворе путем измерения количества электричества, необходимого для полного превращения анализируемого вещества в ходе его электролитического восстановления (окисления) при условии 100 %-ного выхода по току (эффективности тока генерации). Если электролиз проводят в условиях, когда смешение катодных и анодных продуктов исключено, то все количество электричества, прошедшее через раствор в процессе электролиза, расходуется только на окисление (анодная реакция) или восстановление (катодная реакция) единственного вещества. Количество электричества, израсходованного за время протекания реакции до полного разложения реагирующего вещества, определяют содержание этого вещества, основываясь на известных законах электролиза. Единица количества электричества - кулон (Кл, А-с)' и дала название методу анализа -кулонометрия.[28] Соотношение между количеством электричества и массой превратившегося в ходе электролиза вещества установлено законом Фарадея:
т = Mit/nF,
где т - масса прореагировавшего (определяемого) вещества, г; М - его молярная масса, г/моль; i - сила тока в цепи, А; т - время электролиза, с; п - число электронов, принимающих участие в реакции с 1 молекулой определяемого вещества; F-постоянная Фарадея, равная 96500 Кл/моль (А-с/моль). Значение потенциала рабочего электрода, которое надо задать для определения интересующего вещества, можно установить разными способами. Электродный потенциал Е для любой концентрации электродноактивных веществ можно вычислить с помощью уравнения Нернста, записанного в форме:
E Ox/Red = E0* + RT/(nF)ln(COx/CRed) 0* - формальный потенциал оксред системы. На практике для проведения электролиза потенциал рабочего электрода устанавливают более отрицательным (при восстановлении) или более положительным (при окислении) относительно формального значения потенциала. Величина сдвига Е определяется желаемой степенью завершенности электрохимического превращения вещества. В этом случае сила тока, протекающего при электролизе в стационарных условиях, определяется уравнением:
I = I д. пр.[1-exp (nF/(RT)DE)] д. пр -сила предельного тока, который может быть получен при данной концентрации вещества в случае диффузионного контроля скорости переноса вещества к электроду. Можно вести электролиз на предельной силе. Поскольку сила предельного тока пропорциональна концентрации, то по мере протекания электролиза регистрируемая сила тока будет падать (Е = const по условию) в соответствии с убылью концентрации вещества в растворе. Падение силы тока описывается уравнением:
I=I exp(-kt)
I -сила тока в начальный момент времени электролиза, А; k - константа; t время, прошедшее от начала электролиза до момента измерения тока, с. Последнее уравнение позволяет вычислить время электролиза, необходимое для разложения анализируемого вещества с любой полнотой, если известна константа k. Значение k зависит от коэффициента диффузии электроактивного вещества D, площади поверхности рабочего электрода А, объёма анализируемого раствора V и толщины диффузионного слоя по уравнению =DA/(V .
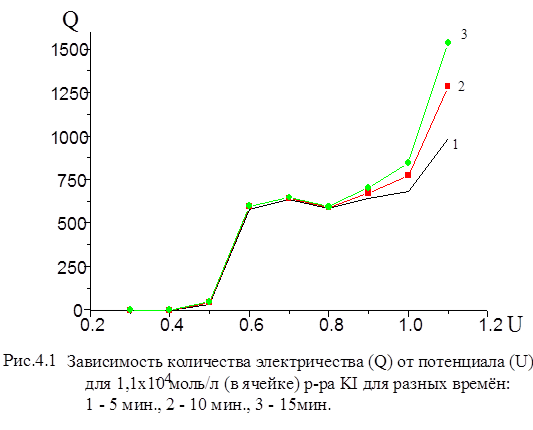
В полулогарифмических координатах (lgI - t ) эта зависимость имеет вид прямой линии. Экспериментальная кривая зависимости, I от имеет более сложный вид. Искажения на начальном участке связаны с токами заряжения двойного электрического слоя (ДЭС), на конечном - с влиянием разложения неизбежных примесей. Имеются и другие причины нарушения хода кривой.[28] Инструментальные погрешности кулонометрической установки с контролируемым потенциалом можно разделить на две основные группы. Погрешности I группы - погрешности коэффициента передачи процесса “масса определяемого вещества ® отсчет прибора”. Основными составляющими погрешности являются: 22.Величина выхода по току электролитической ячейки (отличие от 100%). Эта составляющая полностью определяется конструкцией ячейки (отсутствие потерь вещества на стенках и диафрагмах, устранение брызг и “карманов” и т.п.) и может быть сведена к величине менее 0, 01%; 23.Коэффициент завершенности процесса электролиза. Погрешность величины коэффициента завершенности определяется факторами: · погрешность установки потенциала рабочего электрода. При потенциалах, отличающихся от формального на ±180 мВ, неточность установки потенциала в 1мВ изменяет величину коэффициента завершенности на 0, 004%; · нестабильность поддержания потенциала вследствие пульсаций тока электролиза и температурной нестабильности потенциала в процессе анализа. При оптимальной конструкции потенциостата этот вид погрешности может быть снижен до 0, 001-0, 003%; · изменение степени завершенности процесса электролиза при изменении постоянной времени ячейки из-за изменения величины не учитываемой части электролизного тока. При соотношении продолжительности электролиза T и постоянной времени ячейки τ, равном T/τ =12 (T=600c, τ =50c), изменение постоянной времени на 15% приводит к дополнительной погрешности, равной 0, 003%, а изменение на 30% приводит к погрешности 0, 02%. 1. Нелинейность интегратора тока электролиза изменяет общую величину интеграла. В большинстве интеграторов нелинейность увеличивается обратно пропорционально величине интегрируемого сигнала. Особой разновидностью нелинейности является погрешность дискретности цифровых интеграторов, которая также растет обратно пропорционально величине тока и должна обязательно учитываться. 2. Коэффициент передачи интегратора (цена единицы отсчета)- прямо влияет на погрешность кулонометрического анализа. Составляющими этой погрешности являются: · погрешность, определяемая временной нестабильностью коэффициента передачи интегратора. Данная составляющая может быть существенно снижена периодической градуировкой; · погрешность вследствие изменения коэффициента передачи интегратора при изменении температуры. Эта погрешность, в основном, определяется стабильностью измерительного резистора. Суммарная величина этой погрешности может быть доведена до 0, 0003-0, 0006% на 1°С и может быть дополнительно снижена термостатированием помещения и отдельных узлов кулонометрической установки. Погрешности II группы- погрешности градуировки кулонометрической установки. К ним можно отнести: 1. Погрешности градуировки задатчика потенциала. Практически полностью определяется погрешностью вольтметра постоянного тока, используемого при градуировке, и числом разрядов цифроаналогового преобразователя, синтезирующего выходное напряжение задатчика. При использовании лучших моделей вольтметров погрешность градуировки не превышает 0, 1-0, 2 мВ. 2. Погрешность градуировки интегратора (определение цены единицы отсчета). Определяется следующими факторами: · погрешность задания эталонного тока (тока градуировки). Определяется погрешностями вольтметра постоянного тока и катушки образцового сопротивления, по которым вычисляется величина тока. Реально эта величина может быть снижена до 0, 01%. Суммарная величина инструментальной погрешности может быть доведена до 0, 02% (при изменении температуры на 1°С) или 0, 04% (при изменении температуры на 10°С). При этом определяющими факторами являются величина выхода по току электролитической ячейки, нелинейность интегратора тока электролиза и погрешность градуировки интегратора (цены единицы отсчета). На рис. 4.1 приведена измеренная нами зависимость количества электричества от потенциала рабочего электрода для иодид-иона, из которой видно, что в области потенциалов U = 0, 6 - 0, 8 В выход йода по току в течении 10 - 15 мин приближается к 99 ±1%.
Точное значение мы не определяли, но из градуировочной зависимости количества электричества от концентрации иодид-ионов, при решении обратной задачи-нахождение концентрации по величине Q - определили, что погрешность не превышает 1%. На рис. 4.2 приведена аналогичная зависимость для бромид - иона. В области потенциалов 1, 2 -1, 3 В наблюдается “плато”.
Рис. 4.2 Зависимость количества электричества (Q) от потенциала (U) для 1, 1´ 10-4 моль/л раствора KBr (в ячейке).
При измерении временных зависимостей при U = 1, 2 В Q при фиксированной концентрации бромид-иона проявляется существенное уменьшение скорости электрохимической реакции (рис.4.3, нижняя кривая). Попытка измерить градуировочную зависимость Q - C при U = 1, 2 В удовлетворительных результатов не дала. По нашему мнению, это связано с возможным частичным окислением Pt на рабочем электроде В связи с выше сказанным мы были вынуждены уменьшить рабочий потенциал для выделения брома до U = 1, 1 В. При измерении количества электричества для бромид-иона было замечено, что в присутствии следовых количеств иодид-иона наблюдается ускорение электрохимического окисления бромида до брома (рис. 3, верхняя кривая).
Рис. 4.3 Окисление бромид-ионов с добавкой и без добавки иодид-ионов при разных потенциалах
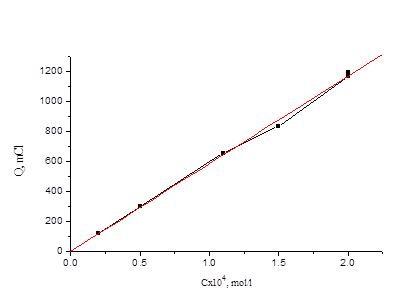
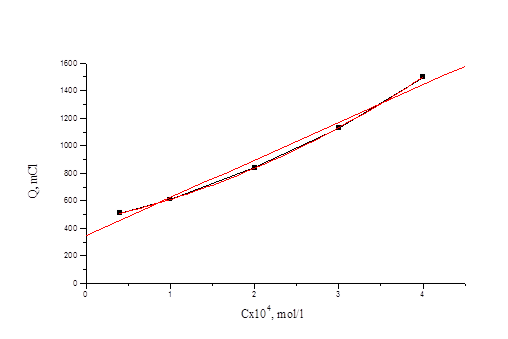
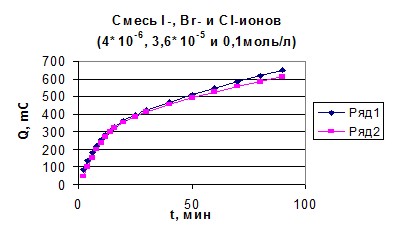
Из рис. 4.1 видно, что при потенциале U = 1, 1 В для иодида характерно более глубокое окисление. Скорость реакции окисления иодида достаточно высокая, а продукты могут взаимодействовать с бромид-ионами, что в результате и ускоряет реакцию окисления бромид-иона. На рис. 3 (верхняя кривая) - зависимость Q от времени для бромид-ионов в присутствии иодид-ионов с концентрацией, соответствующей 0, 1 % от концентрации бромида. Кулонометрическое определение модельной смеси I и Br заключается в следующем: раствор, содержащий аликвоты растворов бромид-и иодид-ионов помещаем в ячейку и ведем электролиз при потенциале U = +0, 75 В в течение 15 минут. Затем данный раствор помещаем в делительную воронку и четырехкратным экстрагированием четыреххлористым углеродом извлекаем выделившийся йод. Аликвоту оставшегося раствора переносим в мерную колбу, разбавляем и проводим электролиз данного раствора при потенциале U = 1, 1 В. По количеству выделившегося электричества рассчитываем концентрации иодид-и бромид-ионов. На рис. 4.4, 4.5 приведены градуировочные зависимости (Q от с) для иодид-иона и для бромид-иона в присутствии небольших (“остаточных”) количеств иодида.
Рис. 4.4 Зависимость количества электричества от концентрации иодид-иона для растворов смеси иодид- и бромид-ионов.
Рис. 4.5 Зависимость количества электричества от концентрации бромид-иона для растворов смеси иодид- и бромид-ионов.
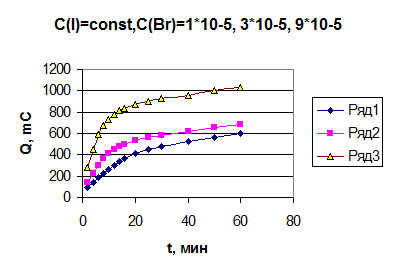
Как видно, строгая линейная зависимость выполняется только для иодида, а для бромида зависимость можно апроксимировать линией. При числе градуировочных растворов-5 в исследованном нами диапазоне концентраций можно использовать и нелинейный градуировочный график. Продолжая исследования, в первую очередь, были получены зависимости количества электричества от времени для йодид-ионов при потенциале выделения брома (U=1, 1 В). При данном потенциале для йодида характерно более глубокое окисление предположительно до йодата. Исходя из этого, мы рассчитывали, какое количества электричества при этом должно выделится. Из рис. 4 видно, что при потенциале 1, 1 В теоретический выход при больших концентрациях (порядка 10-4 моль/л) достигается за длительный промежуток времени, т.е. электрохимическое окисление идет медленно. Зная о том, что незначительные (“остаточные”) добавки иодида к раствору бромида ускоряют реакцию выделения брома, был поставлен ряд опытов, в которых концентрация бромида была постоянной, а концентрация иодида изменялась в пределах 4 10-6-1 10-4 моль/л. Полученные данные (рис. 5 и рис. 6) показали, что теоретически рассчитанное количество электричества достигается за более короткий промежуток времени. Но сходимость результатов нескольких опытов была невысока. Следующим этапом данной работы стало выяснение причин, по которым результаты значительно отличались друг от друга. Предполагалось, что на поверхности платиновых электродов адсорбировались продукты окисления. Проведя ряд экспериментов, в которых в процессе электролиза проводили очистку поверхности рабочего электрода азотной кислотой (1: 1) в течение 15 мин и далее проводили электролиз. Эта очистка поверхности рабочего электрода желаемого результата не дала, так как после очистки поверхности скорость электролиза не увеличивалась, как мы того ожидали (рис. 5). Затем было предложено добавлять в рабочий раствор хлорид-ионы в концентрации, превышающей концентрацию бромид-ионов в 100-150 раз. Предполагалось, что такое количество хлорид-ионов будет препятствовать связыванию иодид- и бромид-ионов в комплексы. Сходимость результатов улучшилась, но не настолько, как нам хотелось. Следующим предположением стало то, что на ход электролиза может влияет последовательность сливания реагентов. В начале было решено разбавлять аликвоту хлорной кислотой бидистиллированной водой, так как концентрация кислоты составляет 1, 03 моль/л, а при такой концентрации она является сильным окислителем. Если после вливания кислоты в колбу первым добавить иодид, то может произойти его окисление до йода, который, адсорбируясь на поверхности рабочего электрода, замедляет скорость электролиза и изменяет продолжительность процесса. По результатам нескольких последовательных экспериментов было установлено, что реагенты должны сливаться в такой последовательности: сначала вливается хлорная кислота, которая разбавляется небольшим количеством бидистиллированной воды, затем добавляли хлорид натрия (100-150-кратный избыток по отношению к бромиду), бромид и только затем иодид. В дальнейшей работе данный порядок сохранялся, так как сходимость результатов анализа улучшилась (рис. 4.6)
. рис. 4.6 Зависимость количества электричества от времени
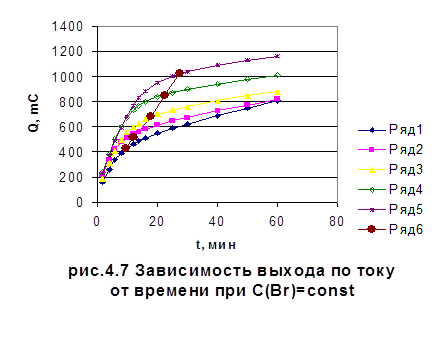
Также в работе была исследована зависимость количества электричества от времени при различных температурах. Результаты показали, что при 200С электролиз протекает медленнее, чем при 250С, а разница в скорости электролиза при 250С и 300С незначительна. Из этого сделали вывод, что электролиз предпочтительнее проводить при 250С, так как незначительное повышение температуры в процессе электролиза на 1-2К не влияет на скорость процесса, и, следовательно, на сходимость результатов (рис. 4.7).
рис. 4.7 Зависимость количества электричества от времени для различных концентраций бромида при постоянной концентрации иодида
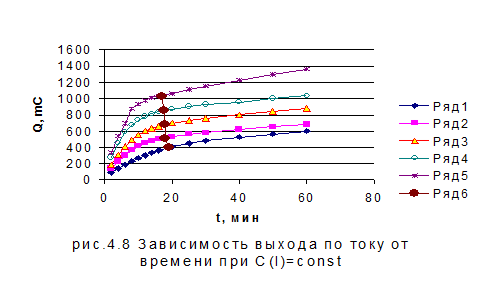
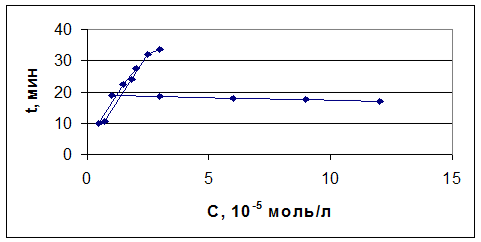
Добившись сходимости результатов, мы смогли начать работать над анализом модельных смесей. В начале мы анализировали рабочие растворы, в которых концентрация иодид-ионов оставалась постоянной, а концентрация бромид-ионов изменялась в пределах 1 10-5-1, 2 10-4 моль/л. В результате были получены зависимости количества электричества от времени. Зависимости времени, соответствующего теоретическому количеству электричества, от концентрации бромид-ионов приведены на рис. 11. При этом время выхода для различных концентраций бромид-ионов колебалась в пределах от 17 до 19 мин. Следующим этапом работы стало проведение электролиза в условиях, когда концентрация бромида выбиралась постоянной, а концентрация иодида варьировалась в пределах 5 10-6 - 3 10-5 моль/л. На рис. 4.8 и рис. 4.9 приведены зависимости количества электричества от времени и зависимости времени, соответствующего теоретическому количеству электричества, от концентрации при потенциале 1, 1 В для смеси иодид- и бромид-ионов. Как видно, при постоянной концентрации бромида теоретический выход по току для иодид-ионов зависит от его концентрации. В то время, как при постоянной концентрации иодида хронометрическая зависимость выражена очень слабо.
- 0, 25, 2- 0, 5, 3- 1, 4-1, 5, 5-2 x 10 -5 моль/л KI 5 - Временная зависимость теоретического выхода по току
- 1, 2- 3, 3- 6, 4-9, 5-12 x 10 -5 моль/л KBr - Временная зависимость теоретического выхода по току Если принять условие, что количество электричества определяется преимущественно концентрацией иодида, то можно реализовать определение иодид- и бромид-ионов из параллельных проб без разделения: При потенциале 0, 75 В определяем иодид (по ранее разработанной методике), а затем при потенциале 1, 1 В проводят измерение суммарного количества электричества для двух ионов. При этом бромид окисляется до брома, а иодид предположительно до иодата. Однако, как видно из временных зависимостей, электрохимическая реакция хотя и замедляется, но не останавливается, а продолжает идти более глубокое окисление. В основе используемой нами методики лежит зависимость времени выхода по току от теоретически ожидаемой величины. Проводя измерения концентрации иодида при потенциале 0, 75 В и используя временную зависимость, представленную на рис. 4.9, оценивают время электролиза смеси при потенциале 1, 1 В.
рис. 17 Зависимость времени, соответствующего теоретическому количеству электричества, от концентрации
(1) - при постоянной концентрации бромида (С=2 х 10-5, 6 х 10-5 моль/л) (2) - при постоянной концентрации иодида( С=1 х 10-5 моль/л) Анализ проводили для модельных смесей с заранее заданными концентрациями иодида и бромида. Для различных соотношений иодид- и бромид-ионов время, при котором определялось суммарное количества электричества также различалось. Концентрация иодида определяется по количеству электричества, выделившегося при потенциале 0, 75 В, а концентрация бромида (в моль/л) в ячейке определяется по формуле:
где Qt, 1.1 - количество электричества, пошедшего на электролиз смеси при потенциале рабочего электрода 1.1 В в течение времени, соответствующего предварительно найденной концентрации иодида; Q0.75 - количество электричества, пошедшего на электролиз смеси при потенциале рабочего электрода 0.75 В в течение 10 мин.; F - постоянная Фарадея; V - объем ячейки в л. В таблице 4.1 приведены результаты определения иодида и бромида в модельных смесях. Определение иодид-иона возможно с достаточно высокой сходимостью. Относительная погрешность определения иодида составляет 2-4%, а бромида 2-10%.
Таблица 4.1
Выводы
1. В периодической и монографической научной литературе отсутствует информация об одновременном определении электроактивных ионов с близкими потенциалами окисления/восстановления с использованием метода потенциостатической кулонометрии. 2. Показано, что в присутствии остаточного количества иодид иона скорость реакции электрохимического окисления бромид-иона возрастает. 3. Показано, что градуировочная зависимость количество электричества - концентрация бромид-иона в присутствии остаточного количества иодид-иона близка к линейной и не проходит через начало координат. 4. Разработан вариант кулонометрического определения иодид- и бромид-иона при совместном присутствии. 5. Измерены зависимости выхода по току продуктов реакции окисления йодид- и бромид-ионов при потенциале 1, 1 В относительно насыщенного хлоридсеребряного электрода. Время выхода по току линейно зависит от концентрации бромид- и йодид-ионов в растворе. 6. Разработан вариант методики одновременного определения бромид- и йодид-ионов при разных потенциалах из параллельных проб. Погрешности определения для йодид-ионов составляют 2-4 %, а для бромид-ионов от2 до 10 %. Список ЛИТЕРАТУРЫ
7. Уильямс У.Дж. Определение анионов: Справочник. Пер. с англ. - М.: Химия, 1982- 624 с. 8. MacKellar Williams J., Droege M., Anderson B.J., Tallman D.E. A microprocessor controlled coulonometric titrator system for the determination of chloride ion // Chem., Biomed., and Environ. Instrum. - 1982. - V.12. - №1. - PP.39-54. - Цит. по РЖХим: 1982, 18Г72. 9. Montiel A., Dupont J. J. Dosage des ions chlorures dans les eaux par microcoulometric // Trib.CEBEDEAU. - 1974. - V.27. - №362. - Р.27-30. - Цит. по РЖХим: 1974, 16И199. 10.Островидов Е. А. Кулонометрическое определение хлорид-ионов с помощью проточного индикаторного электрода // Журн. аналит. химии.-1978. - Т.33. - №10. - С.1987-1990. 11.Hu Xiao-Jing, Bai Ling. Determination of chlorine ion content in magnesite by microcoulometric method // Gaodeng xuexiao huaxun xuebao = Chem. J. Chin. Univ.- 1999.-20, Suppl.-C.244. - Цит. по РЖХим: 2000, 00.11-19Г190. 12.Семенов И.А., Комарова Н.И., Никулина А. Г. Кулонометрическое определение хлоридов с предварительным окислением свободного хлора висмутатным методом // Журн. аналит. химии. - 1981. - Т.36. - №6. - С.1221-1223.. 13.Hon Pink-Kay, Townshend A. An improved spectrophotometric determination of chloride via chromil chloride formation // Anal. Сhim. Аcta. - 1980. - V.15. - PP.395-399. 14.Путилина О. Н. Способ определения хлоридов / Донец. НИИ гигиены труда и професс. - № 4633468/26; заявл. 9.1.89; опубл. 23.11.91, бюл. № 43 - Цит. по РЖХим: 1992, 20Г188П. 15.Taniguchi H., Yoshida T., Adachi Y., Nakano S. Fluorometric determination of chloride with 2-(5-Nitro-2-furil)benzotiasole // Chem. and Pharm. Bull. - 1980. -V.28. - № 10. - PP. 2909-2914. - Цит. по РЖХим: 1981, 9Г128. 16.Fluorometric analysis of chloride ion and chemical sensor therefor: Пат. 5691205, США, МПК6 G 01 № 33/00/ Kawabata Y., Toge Y., Canon KK. - №493346; заявл. 21.06.1995. Опубл. 25.11.1997- Цит. по РЖХим: 2000, 00.19-19Г119П. 17.Соловьев Е. А., Комлева В. И. Способ определения хлоридов. - А.С. 1191824, СССР. Заявл.13.01.84, опубл. в В. И., 1985, № 42. МКИ G 01 № 31/22, 21/76. 18.Spectrophotometric determination of trace bromide by catalytic effect on the tetrabase-chloramine T reaction / Tomiyasu Tokashi, Taga Yoshiko, Sakamoto Hayao, Yonehara Norinobu // Anal. Chim. Acta.-1996.-319, № 1-2.- PP.199-204. 19.Oosting M., Reijnders H.F.R. Spectrophotometric determination of bromide in aqueous solutions // Fresenius Z. Anal. Chem. - 1980. - V.301. - N1. - PP.28-29. 20.Кочетова Т.М., Новикова Т.М. Определение микроколичеств бромида в воде // Хим. и техн. воды. - 1981. - Т.3. - №1. - С.60. 21.Vohiyama Chunichi, Muto Giichi. Determination of bromination number by controlled-potential coulometry with controlled-current coulometric generation of bromine // Anal. Chem. - 1984. - V.56. - N.13. - PP.2408-2410. Цит. по РЖХим: 1985, 12Г37. 22.Verfahren zur schnellen Bromid bestimmung: Пат. 273506 ГДР, МКИ4 G01 N27/42 /Johl W., Sickel H., Streitz A.; VEB Komninat Kali, Schacht II. - N3172294. - Заявл. 28.06.88. Цит. по РЖХим: 1990, 15Г273П 23.Островидов Е.А., Уланова Н.Н. Определение микроколичеств бромид-иона проточным кулонометрическим методом // Заводск. лаб. - 1979. - Т.45. - №6. - С.491-492. 24.Гайдук О.В., Панталер Р.П., Бланк А.Б. Фотометрическое определение микрограммовых количеств иодидов в природных и сточных водах // Вестн. Харьк. ун-та. - 1999. - №454. - С.111-114. 25.Agrawal Omi, Sunita G., Gupta V.K. A sensitive colorimetric method for the micro determination of iodine in marine water // Talanta. - 1999. - V.49. - N.4. - PP.923-928. Цит. по РЖХим: 2001, 0101-19Г218. 26.Rezaoi B. Flow injection determination of trace amounts of iodide with spectrophotometric detection by its catalytic effect on the 4, 4’-bis-(dimethylamino)-diphenylmethane - chloramine T reaction // Anal. Lett. - 2000. - V.33. - N12. - PP.2553-2562. - Цит. по РЖХим.: 2001, 01.01 - 19Г144. 27.Борисова-Пангарова Р. Потенциостатично кулонометрично определяне на малки количества йодни йони чрез окисление върху платинов электрод // Годишник висш. хим.-технолочен институт - София. - 1978. - Т.24. - №1. - С.119-126. - Цит. по РЖХим.: 1981, 23Г175. 28.Zhang Aimei, Wang Shuhao, Du Lingyun, Cui Hui. Simultaneous determination of micro bromide and iodide by kinetic spectrophotomeric method // Anal. Lett.- 2000. - V.33. - N.11. - PP.2321-2331. - Цит. по РЖХим: 2001, 01.01 - 19Г145. 29.Александрова Т.П., Клетеник Ю.Б. Инверсионная вольтамперометрия бромид- и иодид-ионов на обновляемом серебряном электроде // Журн. аналит. химии. - 2000. - Т.55. - №6. - С.655-658. 30.Zhu Chu, Bright F.V., Hieftje G.M. Simultaneous determination of bromide and iodide ions with a multiple fiber-optic fluorescence sensor / Appl. Spectrosc.-1990. - V.44. - N.1. - PP.59-63. - Цит. по РЖХим: 1990, 18Г206. 31.Norinobu Y., Tetsuya Y., Takashi T., Hayao S. Simultaneous spectrophotomeric determination of traces of bromide and iodide based on their catalytic effects on pyrocatechol violet - hydrogen peroxide reaction // Anal. Sci. - 1989. - V.5. - N.2. - PP.175-179. - Цит. по РЖХим: 1989, 20Г215. 32.Akstinat M.H., Rott C. Coulometrische Bestimmung geringer Halogenidkonzentrationen in anorganischen Bindemitteln und mineralischen Rochstoffen // Zem.-Kalk-Gips. - 1988. - 41. - N.3. - PP.138-143. - Цит. по РЖХим: 1988, 19M269. 33.Костромин А.И., Вагизова А.С., Бадретдинова Г.З., Абдуллин И.Ф. Применение электрогенерированного иода (I) для последовательного определения галогенидов // Журн. аналит. химии. - 1985. - Т.40. - №12. - С.2204-2207. 34.Агасян П.К., Хамракулов П.К. Кулонометрический метод анализа. - М.: Химия, 1984. - 168 с. 35.Могилевский А.Н. Прецизионная кулонометрия при контролируемом потенциале. Инструментальные погрешности. -Электрохимические методы анализа (ЭМА-99).Тезисы докладов. V Всероссийская конференция с участием стран СНГ 6-8 декабря 1999 г. - М.: 1999. - с.154-156. |
|||||||||||||||||||||||||||||||||||||||
Последнее изменение этой страницы: 2020-02-17; Просмотров: 104; Нарушение авторского права страницы
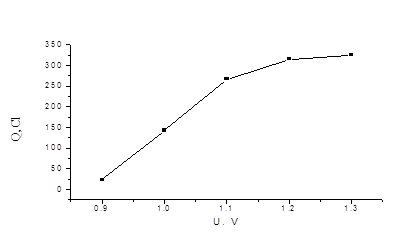

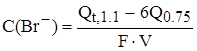 ,
,