
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
ПРИЛОЖЕНИЯ ПЕРВОГО НАЧАЛА ТЕРМОДИНАМИКИ К ХИМИЧЕСКИМ ПРОЦЕССАМСтр 1 из 16Следующая ⇒
ВВЕДЕНИЕ Предметом физической химии является объяснение химических явлений на основе более общих законов физики. Физическая химия рассматривает две основные группы вопросов: 1. Изучение строения и свойств вещества и составляющих его частиц; 2. Изучение процессов взаимодействия веществ. В курсе физической химии обычно выделяют несколько разделов. Строение вещества. В этот раздел входят учение о строении атомов и молекул и учение об агрегатных состояниях вещества. Изучение строение вещества необходимо для выяснения важнейших вопросов об образовании молекул из атомов, о природе химической связи, о строении и взаимодействии молекул. Именно в этой своей части физическая химия очень тесно переплетается со всеми направлениями современной химии, поскольку изучение химических свойств вещества вне связи со строением атомов и молекул на современном уровне невозможно. Химическая термодинамика изучает энергетические эффекты химических процессов; позволяет определить возможность, направление и глубину протекания химического процесса в конкретных условиях. Химическая кинетика. В этом разделе физической химии изучается скорость и механизм протекания химических процессов в различных средах при различных условиях. Учение о растворах рассматривает процессы образования растворов, их внутреннюю структуру и важнейшие свойства, зависимость структуры и свойств от природы компонентов раствора. Электрохимия изучает особенности свойств растворов электролитов, явления электропроводности, электролиза, коррозии, работу гальванических элементов. Коллоидная химия изучает поверхностные явления и свойства мелкодисперсных гетерогенных систем. Все разделы физической химии объединяет единая основа – общие законы природы, которые применимы к любым процессам и любым системам, независимо от их строения. ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА Термодинамика – наука о взаимопревращениях различных форм энергии и законах этих превращений. Термодинамика базируется только на экспериментально обнаруженных объективных закономерностях, выраженных в двух основных началах термодинамики. Термодинамика изучает: 1. Переходы энергии из одной формы в другую, от одной части системы к другой; 2. Энергетические эффекты, сопровождающие различные физические и химические процессы и зависимость их от условий протекания данных процессов; 3. Возможность, направление и пределы самопроизвольного протекания процессов в рассматриваемых условиях. Необходимо отметить, что классическая термодинамика имеет следующие ограничения: 1. Термодинамика не рассматривает внутреннее строение тел и механизм протекающих в них процессов; 2. Классическая термодинамика изучает только макроскопические системы; 3. В термодинамике отсутствует понятие " время". ОСНОВНЫЕ ПОНЯТИЯ ТЕРМОДИНАМИКИ Термодинамическая система – тело или группа тел, находящихся во взаимодействии, мысленно или реально обособленные от окружающей среды. Гомогенная система – система, внутри которой нет поверхностей, разделяющих отличающиеся по свойствам части системы (фазы). Гетерогенная система – система, внутри которой присутствуют поверхности, разделяющие отличающиеся по свойствам части системы. Фаза – совокупность гомогенных частей гетерогенной системы, одинаковых по физическим и химическим свойствам, отделённая от других частей системы видимыми поверхностями раздела. Изолированная система – система, которая не обменивается с окружающей средой ни веществом, ни энергией. Закрытая система – система, которая обменивается с окружающей средой энергией, но не обменивается веществом. Открытая система – система, которая обменивается с окружающей средой и веществом, и энергией. Совокупность всех физических и химических свойств системы характеризует её термодинамическое состояние. Все величины, характеризующие какое-либо макроскопическое свойство рассматриваемой системы – параметры состояния. Опытным путем установлено, что для однозначной характеристики данной системы необходимо использовать некоторое число параметров, называемых независимыми; все остальные параметры рассматриваются как функции независимых параметров. В качестве независимых параметров состояния обычно выбирают параметры, поддающиеся непосредственному измерению, например температуру, давление, концентрацию и т.д. Всякое изменение термодинамического состояния системы (изменения хотя бы одного параметра состояния) есть термодинамический процесс. Обратимый процесс – процесс, допускающий возможность возвращения системы в исходное состояние без того, чтобы в окружающей среде остались какие-либо изменения. Равновесный процесс – процесс, при котором система проходит через непрерывный ряд равновесных состояний. Энергия - мера способности системы совершать работу; общая качественная мера движения и взаимодействия материи. Энергия является неотъемлемым свойством материи. Различают потенциальную энергию, обусловленную положением тела в поле некоторых сил, и кинетическую энергию, обусловленную изменением положения тела в пространстве. Внутренняя энергия системы – сумма кинетической и потенциальной энергии всех частиц, составляющих систему. Можно также определить внутреннюю энергию системы как её полную энергию за вычетом кинетической и потенциальной энергии системы как целого. Формы перехода энергии от одной системы к другой могут быть разбиты на две группы. В первую группу входит только одна форма перехода движения путем хаотических столкновений молекул двух соприкасающихся тел, т.е. путём теплопроводности (и одновременно путём излучения). Мерой передаваемого таким способом движения является теплота. Теплота есть форма передачи энергии путём неупорядоченного движения молекул. Во вторую группу включаются различные формы перехода движения, общей чертой которых является перемещение масс, охватывающих очень большие числа молекул (т.е. макроскопических масс), под действием каких-либо сил. Таковы поднятие тел в поле тяготения, переход некоторого количества электричества от большего электростатического потенциала к меньшему, расширение газа, находящегося под давлением и др. Общей мерой передаваемого такими способами движения является работа – форма передачи энергии путём упорядоченного движения частиц. Теплота и работа характеризуют качественно и количественно две различные формы передачи движения от данной части материального мира к другой. Теплота и работа не могут содержаться в теле. Теплота и работа возникают только тогда, когда возникает процесс, и характеризуют только процесс. В статических условиях теплота и работа не существуют. Различие между теплотой и работой, принимаемое термодинамикой как исходное положение, и противопоставление теплоты работе имеет смысл только для тел, состоящих из множества молекул, т.к. для одной молекулы или для совокупности немногих молекул понятия теплоты и работы теряют смысл. Поэтому термодинамика рассматривает лишь тела, состоящие из большого числа молекул, т.е. так называемые макроскопические системы.
ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ Первое начало термодинамики представляет собой закон сохранения энергии, один из всеобщих законов природы (наряду с законами сохранения импульса, заряда и симметрии): Энергия неуничтожаема и несотворяема; она может только переходить из одной формы в другую в эквивалентных соотношениях. Первое начало термодинамики представляет собой постулат – оно не может быть доказано логическим путем или выведено из каких-либо более общих положений. Истинность этого постулата подтверждается тем, что ни одно из его следствий не находится в противоречии с опытом. Приведем еще некоторые формулировки первого начала термодинамики: Полная энергия изолированной системы постоянна; Невозможен вечный двигатель первого рода (двигатель, совершающий работу без затраты энергии). Первое начало термодинамики устанавливает соотношение между теплотой Q, работой А и изменением внутренней энергии системы Δ U: Изменение внутренней энергии системы равно количеству сообщенной системе теплоты минус количество работы, совершенной системой против внешних сил.
Уравнение (I.1) является математической записью 1-го начала термодинамики для конечного, уравнение (I.2) – для бесконечно малого изменения состояния системы. Внутренняя энергия является функцией состояния; это означает, что изменение внутренней энергии Δ U не зависит от пути перехода системы из состояния 1 в состояние 2 и равно разности величин внутренней энергии U2 и U1 в этих состояниях:
Следует отметить, что определить абсолютное значение внутренней энергии системы невозможно; термодинамику интересует лишь изменение внутренней энергии в ходе какого-либо процесса. Рассмотрим приложение первого начала термодинамики для определения работы, совершаемой системой при различных термодинамических процессах (мы будем рассматривать простейший случай – работу расширения идеального газа). Изохорный процесс (V = const; Δ V = 0). Поскольку работа расширения равна произведению давления и изменения объема, для изохорного процесса получаем:
Изотермический процесс (Т = const). Из уравнения состояния одного моля идеального газа получаем:
Отсюда: Проинтегрировав выражение (I.6) от V1 до V2, получим
Изобарный процесс (Р = const).
Подставляя полученные выражения для работы различных процессов в уравнение (I.1), для тепловых эффектов этих процессов получим:
В уравнении (I.12) сгруппируем переменные с одинаковыми индексами. Получаем:
Введем новую функцию состояния системы – энтальпию H, тождественно равную сумме внутренней энергии и произведения давления на объем:
Тогда выражение (I.13) преобразуется к следующему виду:
Т.о., тепловой эффект изобарного процесса равен изменению энтальпии системы. Адиабатический процесс (Q = 0). При адиабатическом процессе работа расширения совершается за счёт уменьшения внутренней энергии газа:
В случае если Cv не зависит от температуры (что справедливо для многих реальных газов), работа, произведённая газом при его адиабатическом расширении, прямо пропорциональна разности температур:
Закон Гесса Как известно, большинство химических реакций сопровождаются выделением (экзотермические реакции) либо поглощением (эндотермические реакции) теплоты. Первое начало термодинамики дает возможность рассчитать тепловой эффект химической реакции при различных условиях её проведения. Тепловой эффект (теплота) химической реакции – количество теплоты, выделившейся либо поглотившейся в ходе реакции. Тепловой эффект относят, как правило, к числу молей прореагировавшего исходного вещества, стехиометрический коэффициент перед которым максимален. Например, реакцию окисления водорода в химической термодинамике записывают в виде: Н2 + 1/2 О2 ––> Н2О и тепловой эффект рассчитывают на 1 моль водорода. Тепловые эффекты, сопровождающие протекание химических реакций, являются предметом одного из разделов химической термодинамики – термохимии. Определим некоторые понятия термохимии. Теплота образования вещества – тепловой эффект реакции образования 1 моля сложного вещества из простых. Теплоты образования простых веществ принимаются равными нулю. Теплота сгорания вещества – тепловой эффект реакции окисления 1 моля вещества в избытке кислорода до высших устойчивых оксидов. Теплота растворения – тепловой эффект процесса растворения 1 моля вещества в бесконечно большом количестве растворителя. Теплота растворения складывается из двух составляющих: теплоты разрушения кристаллической решетки (для твердого вещества) и теплоты сольватации:
Поскольку Δ Нкр.реш всегда положительно (на разрушение кристаллической решетки необходимо затратить энергию), а Δ Нсольв всегда отрицательно, знак Δ Нраств определяется соотношением абсолютных величин Δ Нкр.реш. и Δ Нсольв:
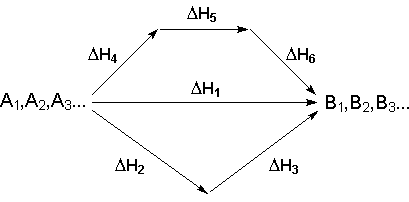
Основным законом термохимии является закон Гесса, являющийся частным случаем первого начала термодинамики: Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания. Выше было показано, что изменение энтальпии Δ Н (тепловой эффект изобарного процесса Qp) и изменение внутренней энергии Δ U(тепловой эффект изохорного процесса Qv) не зависят от пути, по которому система переходит из начального состояния в конечное. Рассмотрим некоторый обобщенный химический процесс превращения исходных веществ А1, А2, А3... в продукты реакции В1, В2, В3..., который может быть осуществлен различными путями в одну или несколько стадий:
Согласно закону Гесса, тепловые эффекты всех этих реакций связаны следующим соотношением:
Практическое значение закона Гесса состоит в том, что он позволяет рассчитывать тепловые эффекты химических процессов. В термохимических расчетах обычно используют ряд следствий из закона Гесса: 1. Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье – Лапласа). 2. Для двух реакций, имеющих одинаковые исходные, но разные конечные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного конечного состояния в другое. С + О2 ––> СО + 1/2 О2 Δ Н 1 С + О2 ––> СО2 Δ Н 2 СО + 1/2 О2 ––> СО2 Δ Н 3
3. Для двух реакций, имеющих одинаковые конечные, но разные исходные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного исходного состояния в другое. С(алмаз) + О2 ––> СО2 Δ Н 1 С(графит) + О2 ––> СО2 Δ Н 2 С(алмаз) ––> С(графит) Δ Н 3
4. Тепловой эффект химической реакции равен разности сумм теплот образования продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты.
5. Тепловой эффект химической реакции равен разности сумм теплот сгорания исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты.
В качестве примера рассмотрим расчет теплового эффекта реакции окисления одного моля глюкозы (теплота образования кислорода по определению равна нулю): С6Н12О6 + 6 О2 ––> 6 СО2 + 6 Н2О
Величины тепловых эффектов химических реакций зависят от условий, в которых проводятся реакции. Поэтому табличные значения теплот различных процессов принято относить к стандартному состоянию – температуре 298 К и давлению 101325 Па (760 мм. рт. ст.; 1 атм.); величины тепловых эффектов при данных условиях называют стандартными тепловыми эффектами и обозначают Δ Н°298 и Δ U°298 соответственно. ТРЕТЬЕ НАЧАЛО ТЕРМОДИНАМИКИ Ранее мы показали, что внутреннюю энергию системы можно условно представить в виде суммы двух величин " свободной" и " связанной" энергии. Возможность рассчитать величину " свободной" энергии, т.е. той части внутренней энергии системы, которую можно превратить в работу, дает тепловая теорема Нернста, называемая также третьим начало термодинамики. Основные положения тепловой теоремы заключаются в следующем: 1. При абсолютном нуле температуры свободная энергия X равна теплоте процесса.
2. При температурах, близких к абсолютному нулю, теплоемкость системы равна нулю. Одной из формулировок третьего начала термодинамики является также постулат Планка: Энтропия идеального кристалла при абсолютном нуле температуры равна нулю. Строго говоря, тепловая теорема Нернста и постулат Планка являются следствиями из второго начала термодинамики; но независимо от этого они имеют очень большое значение, позволяя рассчитывать абсолютную энтропию системы и, следовательно, величину свободной энергии системы. Расчет абсолютной энтропии Рассчитаем изменение энтропии некоторой системы при нагревании её от абсолютного нуля до температуры T при постоянном давлении. Из первого и второго начал термодинамики имеем:
Отсюда:
Учитывая, что ST=0 = 0, получим: При T = 0 любое вещество может находиться только в твердом состоянии. При нагревании вещества возможен его переход в жидкое и затем в газообразное состояние; для фазовых переходов, происходящих в изобарно-изотермических условиях, изменение энтропии равно приведенной теплоте фазового перехода:
Таким образом, нагревание вещества без фазовых переходов сопровождается непрерывным ростом энтропии; при фазовом переходе происходит скачкообразное изменение энтропии. Графическая зависимость энтропии вещества от температуры приведена на рисунке 1.3. Учитывая это, рассчитать абсолютную энтропию любого вещества при любой температуре можно следующим образом:
Рис. 1.3 Зависимость энтропии вещества от температуры.
Поскольку энтропия есть функция состояния, изменение энтропии в ходе химического процесса определяется только видом и состоянием исходных веществ и продуктов реакции и не зависит от пути реакции; оно может быть рассчитано по уравнению (I.67):
Для многих веществ величины абсолютной энтропии в стандартных условиях приведены в справочной литературе. ХИМИЧЕСКОЕ РАВНОВЕСИЕ Как было показано выше, протекание самопроизвольного процесса в термодинамической системе сопровождается уменьшением свободной энергии системы (dG < 0, dF < 0). Очевидно, что рано или поздно (напомним, что понятие " время" в термодинамике отсутствует) система достигнет минимума свободной энергии. Условием минимума некоторой функции Y = f(x) является равенство нулю первой производной и положительный знак второй производной: dY = 0; d2Y > 0. Таким образом, условием термодинамического равновесия в закрытой системе является минимальное значение соответствующего термодинамического потенциала: Изобарно-изотермические (P = const, T = const): Δ G = 0 dG = 0, d2G > 0 Изохорно-изотермические (V = const, T = const): Δ F = 0 dF = 0, d2F > 0 Состояние системы с минимальной свободной энергией есть состояние термодинамического равновесия: Термодинамическим равновесием называется такое термодинамическое состояние системы, которое при постоянстве внешних условий не изменяется во времени, причем эта неизменяемость не обусловлена каким-либо внешним процессом. Учение о равновесных состояниях – один из разделов термодинамики. Далее мы будем рассматривать частный случай термодинамического равновесного состояния – химическое равновесие. Как известно, многие химические реакции являются обратимыми, т.е. могут одновременно протекать в обоих направлениях – прямом и обратном. Если проводить обратимую реакцию в закрытой системе, то через некоторое время система придет в состояние химического равновесия – концентрации всех реагирующих веществ перестанут изменяться во времени. Необходимо отметить, что достижение системой состояния равновесия не означает прекращения процесса; химическое равновесие является динамическим, т.е. соответствует одновременному протеканию процесса в противоположных направлениях с одинаковой скоростью. Химическое равновесие является подвижным – всякое бесконечно малое внешнее воздействие на равновесную систему вызывает бесконечно малое изменение состояния системы; по прекращении внешнего воздействия система возвращается в исходное состояние. Ещё одним важным свойством химического равновесия является то, что система может самопроизвольно прийти в состояние равновесия с двух противоположных сторон. Иначе говоря, любое состояние, смежное с равновесным, является менее устойчивым, и переход в него из состояния равновесия всегда связан с необходимостью затраты работы извне. Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации С, парциальные давления P или мольные доли X реагирующих веществ. Для некоторой реакции
соответствующие константы равновесия выражаются следующим образом:
Константа равновесия есть характерная величина для каждой обратимой химической реакции; величина константы равновесия зависит только от природы реагирующих веществ и температуры. Выражение для константы равновесия для элементарной обратимой реакции может быть выведено из кинетических представлений. Рассмотрим процесс установления равновесия в системе, в которой в начальный момент времени присутствуют только исходные вещества А и В. Скорость прямой реакции V1 в этот момент максимальна, а скорость обратной V2 равна нулю:
По мере уменьшения концентрации исходных веществ растет концентрация продуктов реакции; соответственно, скорость прямой реакции уменьшается, скорость обратной реакции увеличивается. Очевидно, что через некоторое время скорости прямой и обратной реакции сравняются, после чего концентрации реагирующих веществ перестанут изменяться, т.е. установится химическое равновесие. Приняв, что V1 = V2, можно записать:
Т.о., константа равновесия есть отношение констант скорости прямой и обратной реакции. Отсюда вытекает физический смысл константы равновесия: она показывает, во сколько раз скорость прямой реакции больше скорости обратной при данной температуре и концентрациях всех реагирующих веществ, равных 1 моль/л. Теперь рассмотрим (с некоторыми упрощениями) более строгий термодинамический вывод выражения для константы равновесия. Для этого необходимо ввести понятие химический потенциал. Очевидно, что величина свободной энергии системы будет зависеть как от внешних условий (T, P или V), так и от природы и количества веществ, составляющих систему. В случае, если состав системы изменяется во времени (т.е. в системе протекает химическая реакция), необходимо учесть влияние изменения состава на величину свободной энергии системы. Введем в некоторую систему бесконечно малое количество dni молей i-го компонента; это вызовет бесконечно малое изменение термодинамического потенциала системы. Отношение бесконечно малого изменения величины свободной энергии системы к бесконечно малому количеству компонента, внесенному в систему, есть химический потенциал μ i данного компонента в системе:
Химический потенциал компонента связан с его парциальным давлением или концентрацией следующими соотношениями:
Здесь μ ° i – стандартный химический потенциал компонента (Pi = 1 атм., Сi = 1 моль/л.). Очевидно, что изменение свободной энергии системы можно связать с изменением состава системы следующим образом:
Поскольку условием равновесия является минимум свободной энергии системы (dG = 0, dF = 0), можно записать:
В закрытой системе изменение числа молей одного компонента сопровождается эквивалентным изменением числа молей остальных компонентов; т.е., для приведенной выше химической реакции имеет место соотношение:
Отсюда можно получить следующее условие химического равновесия в закрытой системе:
В общем виде условие химического равновесия можно записать следующим образом:
Выражение (I.94) носит название уравнения Гиббса – Дюгема. Подставив в него зависимость химического потенциала от концентрации, получаем:
Поскольку Σ n i μ i = Δ F, а Σ n i μ ° i = Δ F°, получаем:
Для изобарно-изотермического процесса аналогичным образом можно получить:
Полученные нами выражения I.96 – I.97 есть изотерма химическойреакции. Если система находится в состоянии химического равновесия, то изменение термодинамического потенциала равно нулю; получаем:
Здесь сi и рi – равновесные концентрации и парциальные давления исходных веществ и продуктов реакции (в отличие от неравновесных Сi и Рi в уравнениях I.96 – I.97). Поскольку для каждой химической реакции стандартное изменение термодинамического потенциала Δ F° и Δ G° есть строго определенная величина, то произведение равновесных парциальных давлений (концентраций), возведенных в степень, равную стехиометрическому коэффициенту при данном веществе в уравнении химической реакции (стехиометрические коэффициенты при исходных веществах принято считать отрицательными) есть некоторая константа, называемая константой равновесия. Уравнения (I.98, I.99) показывают связь константы равновесия со стандартным изменением свободной энергии в ходе реакции. Уравнение изотермы химической реакции связывает величины реальных концентраций (давлений) реагентов в системе, стандартного изменения термодинамического потенциала в ходе реакции и изменения термодинамического потенциала при переходе из данного состояния системы в равновесное. Знак Δ G (Δ F) определяет возможность самопроизвольного протекания процесса в системе. При этом Δ G° (Δ F°) равно изменению свободной энергии системы при переходе из стандартного состояния (Pi = 1 атм., Сi = 1 моль/л) в равновесное. Уравнение изотермы химической реакции позволяет рассчитать величину Δ G (Δ F) при переходе из любого состояния системы в равновесное, т.е. ответить на вопрос, будет ли химическая реакция протекать самопроизвольно при данных концентрациях Сi (давлениях Рi) реагентов:
Если изменение термодинамического потенциала меньше нуля, процесс в данных условиях будет протекать самопроизвольно. ФАЗОВЫЕ РАВНОВЕСИЯ Вещество при изменении давления и температуры может переходить из одного агрегатного состояния в другое. Эти переходы, совершающиеся при постоянной температуре, называют фазовыми переходами первого рода. Количество теплоты, которое вещество получает из окружающей среды либо отдает окружающей среде при фазовом переходе, есть скрытая теплота фазового перехода λ фп. Если рассматривается гетерогенная система, в которой нет химических взаимодействий, а возможны лишь фазовые переходы, то при постоянстве температуры и давления в системе существует т.н. фазовое равновесие. Фазовое равновесие характеризуется некоторым числом фаз, компонентов и числом степеней термодинамической свободы системы. Компонент – химически однородная составная часть системы, которая может быть выделена из системы и существовать вне её. Число независимых компонентов системы равно числу компонентов минус число возможных химических реакций между ними. Число степеней свободы – число параметров состояния системы, которые могут быть одновременно произвольно изменены в некоторых пределах без изменения числа и природы фаз в системе. Число степеней свободы гетерогенной термодинамической системы, находящейся в состоянии фазового равновесия, определяется правилом фаз, сформулированным Дж. Гиббсом: Число степеней свободы равновесной термодинамической системы С равно числу независимых компонентов системы К минус число фаз Ф плюс число внешних факторов, влияющих на равновесие. Для системы, на которую из внешних факторов влияют только температура и давление, можно записать: С = К – Ф + 2 (I.108) Системы принято классифицировать по числу компонентов (одно-, двухкомпонентные и т.д.), по числу фаз (одно-, двухфазные и т.д.) и числу степеней свободы (инвариантные, моно-, дивариантные и т.д.). Для систем с фазовыми переходами обычно рассматривают графическую зависимость состояния системы от внешних условий – т.н. диаграммы состояния. Анализ диаграмм состояния позволяет определить число фаз в системе, границы их существования, характер взаимодействия компонентов. В основе анализа диаграмм состояния лежат два принципа: принцип непрерывности и принцип соответствия. Согласно принципу непрерывности, при непрерывном изменении параметров состояния все свойства отдельных фаз изменяются также непрерывно; свойства системы в целом изменяются непрерывно до тех пор, пока не изменится число или природа фаз в системе, что приводит к скачкообразному изменению свойств системы. Согласно принципу соответствия, на диаграмме состояния системы каждой фазе соответствует часть плоскости – поле фазы. Линии пересечения плоскостей отвечают равновесию между двумя фазами. Всякая точка на диаграмме состояния (т. н. фигуративная точка) отвечает некоторому состоянию системы с определенными значениями параметров состояния. Рассмотрим и проанализируем диаграмму состояния воды (рис.1.4). Поскольку вода – единственное присутствующее в системе вещество, число независимых компонентов К = 1. В системе возможны три фазовых равновесия: между жидкостью и газом (линия ОА – зависимость давления насыщенного пара воды от температуры), твердым телом и газом (линия ОВ – зависимость давления насыщенного пара надо льдом от температуры), твердым телом и жидкостью (линия ОС – зависимость температуры плавления льда от давления). Три кривые имеют точку пересечения О, называемую тройной точкой воды; тройная точка отвечают равновесию между тремя фазами.
Рис. 1.4. Диаграмма состояния воды
В тройной точке система трехфазна и число степеней свободы равно нулю; три фазы могут находиться в равновесии лишь при строго определенных значениях температуры и давления (для воды тройная точка отвечает состоянию с Р = 6.1 кПа и Т = 273.16 К). Кривая ОВ теоретически продолжается до абсолютного нуля, а кривая давления насыщенного пара над жидкостью ОА заканчивается в критической точке воды (Tкр = 607.46 К, Ркр = 19.5 МПа); выше критической температуры газ и жидкость не могут существовать как отдельные фазы. Кривая ОС в верхней части (при высоких давлениях) изменяет свой наклон (появляются новые кристаллические фазы, плотность которых, в отличие от обычного льда, выше, чем у воды). Внутри каждой из областей диаграммы (АОВ, ВОС, АОС) система однофазна; число степеней свободы системы равно двум (система дивариантна), т.е. можно одновременно изменять и температуру, и давление, не вызывая изменения числа фаз в системе: С = 1 – 1 + 2 = 2 На каждой из линий число фаз в системе равно двум и, согласно правилу фаз, система моновариантна, т.е. для каждого значения температуры имеется только одно значение давления, при котором система двухфазна: С = 1 – 2 + 2 = 1 Влияние давления на температуру фазового перехода описывает уравнение Клаузиуса – Клапейрона:
Здесь Δ Vфп = V2 – V1 есть изменение молярного объема вещества при фазовом переходе (причем V2 относится к состоянию, переход в которое сопровождается поглощением теплоты). Уравнение Клаузиуса – Клапейрона позволяет объяснить наклон кривых равновесия на диаграмме состояния однокомпонентной системы. Для переходов " жидкость – пар" и " твердое вещество – пар" Δ V всегда больше нуля; поэтому кривые на диаграмме состояния, отвечающие этим равновесиям, всегда наклонены вправо (повышение температуры всегда увеличивает давление насыщенного пара). Поскольку молярный объем газа много больше молярного объема того же вещества в жидком или твердом агрегатном состояниях (Vг > > Vж, Vг > > Vт), уравнение (I.109) для частных случаев испарения и возгонки примет следующий вид:
Популярное:
|
Последнее изменение этой страницы: 2016-03-15; Просмотров: 1919; Нарушение авторского права страницы
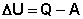 (I.1)
(I.1)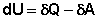 (I.2)
(I.2)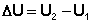 (I.3)
(I.3)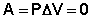 (I.4)
(I.4)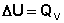 (I.5)
(I.5)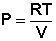 (I.6)
(I.6)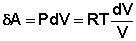 (I.7)
(I.7)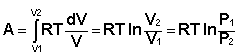 (I.8)
(I.8)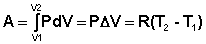 (I.9)
(I.9)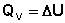 (I.10)
(I.10)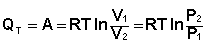 (I.11)
(I.11)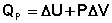 (I.12)
(I.12)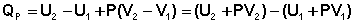 (I.13)
(I.13)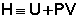
 (I.14)
(I.14)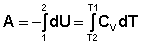 (I.15)
(I.15)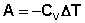 (I.16)
(I.16)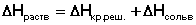

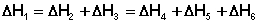 (I.17)
(I.17)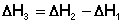 (I.18)
(I.18)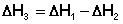 (I.19)
(I.19) (I.20)
(I.20)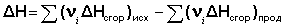 (I.21)
(I.21)
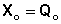 (I.59)
(I.59)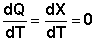 (I.60)
(I.60) (I.61)
(I.61) (I.62)
(I.62)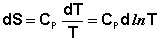 (I.63)
(I.63) (I.64)
(I.64)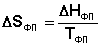 (I.65)
(I.65) (I.66)
(I.66)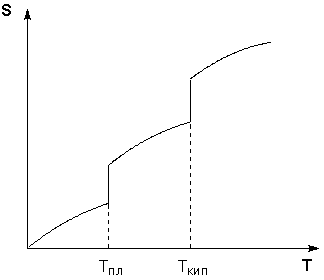
 (I.67)
(I.67)
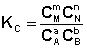 (I.78)
(I.78) 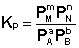 (I.79)
(I.79) 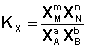 (I.80)
(I.80)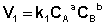 (I.81)
(I.81)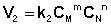 (I.82)
(I.82) (I.83)
(I.83)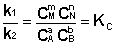 (I.84)
(I.84)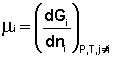 (I.85)
(I.85) 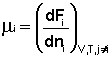 (I.86)
(I.86)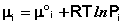 (I.87)
(I.87) 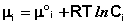 (I.88)
(I.88)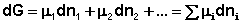 (I.89)
(I.89)  (I.90)
(I.90)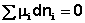 (I.91)
(I.91)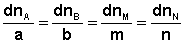 (I.92)
(I.92)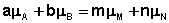 (I.93)
(I.93)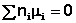 (I.94)
(I.94)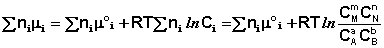 (I.95)
(I.95)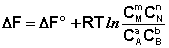 (I.96)
(I.96) (I.97)
(I.97)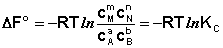 (I.98)
(I.98) 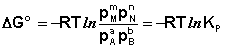 (I.99)
(I.99)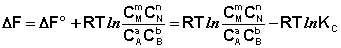 (I.100)
(I.100) (I.101)
(I.101)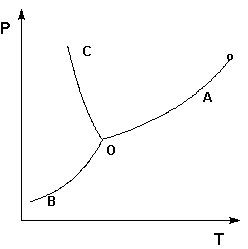
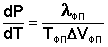 (I.109)
(I.109) (I.110)
(I.110)