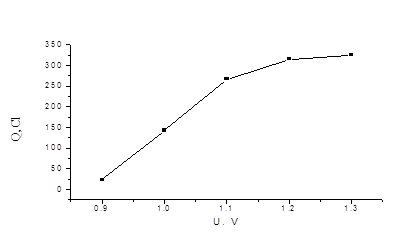|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Определение йодида и бромида в растворах методом потенциостатической кулонометрииСтр 1 из 2Следующая ⇒
Определение йодида и бромида в растворах методом потенциостатической кулонометрии
РЕФЕРАТ
Квалификационная работа на получение ученой степени магистра содержит стр., рис., источников. Объекты исследования: Модельные растворы бромид- и иодид-ионов. Электрохимическое окисление. Цель работы. Провести сопоставление зависимости выхода по току от потенциала для иодид- и бромид-ионов и предложить варианты определения их концентраций в растворах при совместном присутствии. Метод исследования: Потенциостатическая кулонометрия. Результаты: Показано, что в присутствии остаточного количества иодид иона скорость реакции электрохимического окисления бромид-иона возрастает. Разработан вариант последовательного кулонометрического определения иодид- и бромид-иона при совместном присутствии. Измерены зависимости выхода по току продуктов реакции окисления йодид- и бромид-ионов при потенциале 1, 1 В относительно насыщенного хлоридсеребряного электрода. Время выхода по току линейно зависит от концентрации бромид- и йодид-ионов в растворе. Разработан вариант методики одновременного определения бромид- и йодид-ионов при разных потенциалах из параллельных проб. Погрешности определения йодид-ионов составляют 2-4 %, а бромид-ионов 2 - 10 %. Результаты будут использованы для разработки методики определения иодид- и бромид-ионов в сильно минерализованных водах.
СОДЕРЖАНИЕ
Введение . ОБЗОР МЕТОДОВ ОПРЕДЕЛЕНИЯ ГАЛОГЕНИД-ИОНОВ Определение хлорид-ионов Методы определения бромид-ионов Методы определения иодид-ионов Опрелеление галогенид-ионов при совместном присутствии . ОБОСНОВАНИЕ ТЕМЫ РАБОТЫ . ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ Реактивы, растворы и средства измерения Измерение временных зависимостей выхода продуктов при фиксированном потенциале Методика электрохимического окисления иодид-ионов при градуировке и анализе Методика электрохимического окисления бромид-ионов при градуировке и анализе Методика электрохимического окисления смеси бромид- и иодид - ионов при градуировке и анализе Методика электрохимического окисления смеси бромид- и иодид - ионов при градуировке и анализе без отделения йода . ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ Выводы Список литературы
Введение
Метод прямой кулонометрии обладает рядом характерных особенностей, основная из которых - возможность определения концентраций компонентов с высокой точностью. Метод позволяет проводить определения в широком диапазоне концентраций. Однако для получения достоверных результатов необходимо соблюдение следующего условия: электрохимическое превращение вещества должно протекать со 100% выходом по току, то есть должны отсутствовать побочные электрохимические процессы. Задачу определения нескольких компонентов при совместном прохождении реакций окисления/восстановления следует признать актуальной. Даже частичное ее разрешение позволит расширить возможности метода. Одна из систем, на примере которой можно изучить принципиальную возможность одновременного кулонометрического определения - смесь галогенид-ионов, в частности, иодида и бромида. Цель настоящей работы - провести сопоставление зависимости выхода по току от потенциала от иодид- и бромид-ионов и предложить варианты определения их концентраций в растворах при совместном присутствии. Возможные объекты анализа - сильно минерализованные природные воды. бромид иодид градуировка электрохимический 1. ОБЗОР МЕТОДОВ ОПРЕДЕЛЕНИЯ ГАЛОГЕНИД-ИОНОВ
Определение хлорид-ионов
Гравиметрические методы Растворимые хлориды определяют гравиметрически в виде AgCl при добавлении к анализируемой пробе раствора нитрата серебра, осаждение проводят в присутствии разбавленной азотной кислоты. При строгом соблюдении методических указаний можно добиться высокой точности определения хлорида. Этот метод, за исключением современных кулонометрических методов, остаётся основным методом прямого анализа особо чистых хлоридов.[1] Титриметрические методы Прямые титриметрические методы основаны на добавлении небольшого избытка Ag+ после полного осаждения хлорида. Избыточное серебро реагирует с образованием окрашенных комплексов, например, с хроматом. Среди титриметрически методов хорошо известны: метод Фольгарда, метод Мора, метод Фаянса и другие аргентометрические методы. Также к ним относятся меркуриметрические методы, в которых прямое титрование хлоридов ртутью(II), основано на образовании слабо диссоциированого хлорида ртути; комплексонометрический метод, в котором хлорид определяется косвенно с использованием ЭДТА.[1] Спектрофотометрические методы К спектрофотометрическим методам относят метод с применением роданида ртути, который основан на взаимодействии с ним хлорид-ионов с образованием слабо диссоциированого хлорида ртути. Метод предварительного концентрирования хлоридов со осаждением с фосфатом свинца также относят к данным методам. В литературе [7] исследован улучшенный метод спектрофотометрии определения Cl--ионов, основанный на экстракции CCl4 CrOCl2, образующегося при взаимодействии Cl- c K2Cr2O7 в среде H2SO4, реэкстракции CrO2Cl2 цитратным буфером с рН 3, 2, окислении I- до I3- действием CrO2Cl2 и спектрофотометрировании раствора при 288 нм или 351 нм. Относительное стандартное отклонение при определении 10 мкг Cl- составляет 1%. Предложен способ определения Cl- -ионов в воздухе [8], включающий перевод их в комплексные соединения с роданидом ртути железоаммонийными квасцами в кислой среде и последующим фотометрировании. Нефелометрическим методом можно определять хлориды по суспензии AgCl в 50%-ном метаноле. Концентрацию суспензии измеряют при 600 нм.[1] Атомно-абсорбционный метод Косвенный ААС метод, основан на осаждении хлорида с Ag+ и последующем определении или избытка Ag+ в растворе, или Ag+, получающегося после растворения осадка. Другой метод основан на ААС-определении ртути в хлориде фенилртути (II).[1] Потенциометрические методы, ионоселективные электроды Хлориды можно определять с помощью различных потенциометрических методов, начиная с классической потенциометрии и заканчивая современными методами, например: с использованием ионселективных электродов, потенциометрия с наложением постоянного тока, метод концентрационного элемента, потенциометрия до нулевой точки, дифференциальная потенциометрия с использованием серебряного или хлоридсеребряного электрода и др.[1] Хлорид-ионы также определяются с использованием нескольких электродов различного типа. Чаще всего применяют твердые мембранные электроды. Хлоридный электрод обычно используют в паре с каломельным электродом сравнения. Существует и комбинированный хлоридный электрод, в котором в одном корпусе смонтирован и хлоридный ионселективный электрод, и элетрод сравнения.[1]. Кулонометрические методы Кулонометрический анализ используется также для определения хлоридов (см. раздел “Определение бромид-ионов “). В работе [4] изучено кулонометрическое определение хлорид-ионов с помощью металлокерамических проточных пористых Ag-электродов. Этот метод характеризуется относительным стандартным отклонением на уровне 0, 05 при времени единичного определения 15 минут. Предложена микрокулонометрическая методика [5] для определения малых содержаний Cl--иона (0, 01%) в магнезите. Перед анализом пробу (5-10 г) растворяют в 62-68% азотной кислоте в присутствии 30 мл воды и нейтрализуют 30%-ным раствором NaOH. Допустимый разброс напряжений 260-270ли мВ. Поскольку концентрации Br- и I- в магнезите очень малы, их влиянием можно пренебречь. Методика позволяет определить Cl- на уровне 0, 0002-1%. Кулонометрическое титрование электрогенерированным Ag+-ионом применили для определения 1-200 нмоль Cl- в среде уксусная кислота - вода (75: 25). Хотя Ag+-ион легко генерируется из серебряного электрода, присутствие окислителей может затруднить окисление анода независимо от силы тока. Их влияние можно исключить, генерируя Ag+-ион сквозь проницаемую мембрану из Ag2S.[1] При использовании в качестве фонового электролита 0, 5 М раствора NaClO4 и 0, 02 М раствора HClO4, можно определять содержание хлоридов, бромидов и иодидов с высокой чувствительностью и точностью. Однако при титровании смеси галогенидов сталкиваются с процессом со осаждения.[1] В работе [6] показана возможность количественного окисления хлоридов до свободного хлора в кислой среде с помощью висмутата щелочного металла с последующим кулонометрическим определением свободного хлора. Другие методы К другим методам можно отнести амперометрические, полярографические методы, метод хронопотенциометрии. Флуориметрические методы описаны в [9], [10], люминесцентный метод представлен в [11].
1.2 Методы определения бромид-ионов
Спектрофотометрическое определение бромид-ионов Многие спектрофотометрические методы определения хлоридов применимы для бромидов, а в некоторых случаях и для иодидов. Это следующие методы: . Метод с использованием роданида ртути. Сравнимый с ним метод, основанный на применении роданида серебра, применим для определения бромида и иодида в присутствии хлорида. . Метод с использованием дифенилкарбазона - дифенилкарбазида ртути. . Метод с использованием бромидных комплексов Fe111. В этом случае в присутствии бромида появляется полоса поглощения при 420 нм. . Метод, основанный на вытеснении HCN из Hg (CN)2 галогенидом. . Метод, основанный на образовании фенилртуть(II)-галогенидов и их взаимодействии с диэтилдитиокарбаминатом натрия. Некоторые спектрофотометрические методы определения бромида основаны на его предварительном окислении до брома. Такие методы в основном ограниченно селективные. Хлорамин Т часто используют для окисления бромида до брома. После окисления образующийся бром можно определить любым удобным методом. Для определения бромида в водных растворах используют его реакцию с сульфатом палладия с последующим образованием соединений с характерным спектром поглощения в ультрафиолетовой области.[1] В литературе [13] предложен метод определения бромид-ионов, включающий его окисление до Br2 хлорамином Т (І), взаимодействии с флуоресцеином (ІІ) и спектрофотометрическом определении образующегося эозина. Последний, в свою очередь, реагирует с (І) с образованием бесцветного соединения. Поэтому окраска фотометрирующего раствора меняется во времени. Для стандартизации условий определения измерение оптической плотности проводят через 20 минут, прерывая реакцию добавлением избытка тиосульфата натрия. Предел обнаружения 7х10-6 г-ион/л Br2. Калибровочный график прямолинеен до 1, 5х10-4 г-ион/л Br2. Определению не мешают до 1, 5х10-6 г-ион/л I- и 1, 5х10-3 г-ион/л BrO3-. Известна методика [12] спектрофотометрического определения бромид-ионов, основанная на их каталитическом эффекте на реакцию между тетраоснованием [4, 4-бис(диметиламино)дифенилметаном] и хлорамином Т при рН 3, 8. Продукт реакции имеет максимум светопоглощения при 600 нм. В качестве аналитического сигнала использовали светопоглощение продукта в фиксированное время. Диапазон определяемых концентраций равен 2-100 мкг/л. Правильность методики проверена методом стандартных добавок. Аргентометрические методы Для определения бромида можно использовать все три метода, названные методами Фольгарда, Мора и методом абсорбционного индикатора (метод Фаянса). Методы Фольгарда и Мора описаны в [1]. Для метода абсорбционного индикатора эозин (тетрабромфлуоресцеин) является лучшим индикатором, но флуоресцеин также подходит для проведения определения. Бромид можно титровать меркуриметрически, используя в качестве индикаторов нитропруссид, дифенилкарбазид и дифенил-карбазон. Редокс-титриметрические методы определения бромида основанны на окислении его до брома или бромата. Также бромид можно определить, используя церий (IV) или тетраацетат свинца.[1] Потенциометрические методы, ионоселективные электроды Потенциометрические методы, описанные для хлоридов, в значительной степени применимы и для бромидов. В классическом титровании при использовании стандартного раствора AgNO3 в качестве титранта кривая титрования получается очень четкой.[1] В некоторых случаях дифференциальная электролитическая потенциометрия применима для определения бромидов. А использование метода кулонометрии при постоянном токе позволяет определять 10--11 M бромида, при этом реакцию проводят в ячейке емкостью 0, 5 мл в среде 0, 01 М НN03 в смеси 80: 20 метанол - вода.[1] Бромид в присутствии хлорида, иодида и фторида можно оттитровать потенциометрически, используя смешанный титрант AgNO3 -Th(NO3)4. Другие потенциометрические методы, например, с использованием ртути(II), менее применимы для определения бромидов и хлоридов. Для исследований иногда применяется прямое потенциометрическое определение бромида в интервале концентраций 10-1-10-4 М с использованием Ag-AgBr-индикаторного электрода и каломельного электрода сравнения. В качестве редокс-титрантов для потенциометрического определения бромидов рекомендуют также тетраацетат свинца, кобальт(III) и церий(IV). А применение ионоселективных электродов для определения бромидов практически не отличается от использования их для анализа хлоридов.[1] Кулонометрические методы Бромид, так же как хлорид и иодид, можно определять кулонометрическим методом. Обычно используют метод с электрогенерацией иона серебра. Данный метод основан на измерении количества электричества, необходимого для количественного протекания реакции по уравнению + X-- ® AgX + e- X = Cl--, Br--, I--.
Галогенид разряжается на серебряном аноде. Лингейном [1] описан вариант метода, в котором точку эквивалентности определяют потенциометрически. Метод позволяет определять 0, 2-10 мг хлорида, бромида или иодида в 50 мл раствора. Точность метода выше, чем при обычном титровании галогенидов раствором AgNO3 с использованием индикаторов. При этом смесь галогенидов анализируют, проводя последовательный электролиз при подходящем значении потенциала. Так, известно определение 5 - 100 мкг Вг-- в образце, содержащем хлорид. Предложен способ [16] быстрого определения содержания брома в виде Br- в солевых композициях или растворах в присутствии значительных концентраций Cl-. Способ заключается в окислении Br- соединениями Ce (4+) и последующем кулонометрическом определении его концентрации. В научной литературе предложен проточный кулонометрический метод определения бромид-ионов [17] в водных растворах с использованием металлокерамического электрода из серебра. Время определения 15 минут; точность - 4%. Для выполнения определения достаточно 5 мкл анализируемого раствора. Амперометрические методы Разработаны амперометрические методы определения бромидов. Бромид можно определять, используя в качестве титранта Cd(NO3)2 в среде безводной уксусной кислоты. Науке известен полярографический метод определения бромид-ионов. Каталитические методы Из литературных данных известно, что аммоний окисляется гипохлоритом до трихлорамина по следующей реакции: 3 + ЗСlO-- + ЗН+ ® NC13 + ЗН2О
В присутствии бромида гипохлорит окисляет бромид до гипобромита, который в свою очередь окисляет аммоний до азота: СlO-- + Вг--- ® ВгO-- + СI- 3BrO-- + 2NH3 ® N2 + 3H2O + 3Br--
Таким образом, бромид ингибирует реакцию хлорирования аммония до трихлорамина. Это и было использовано в качестве основы каталитического спектрофотометрического метода определения 0, 02 - 1, 20 ррm бромида.[1] Основой других методов явилось каталитическое влияние бромида на окисление иода до иодата CeIV в присутствии К2Сг2О7 в среде HNO3 [1] и ускорение разложения бромата в уксуснокислой среде [14]. Радиохимические методы Бромид и хлорид можно определять, добавляя к анализируемому раствору 203Hg(IO3)2 и измеряя активность образующихся в результате реакции HgBr2 или HgCl2. Нейтронно-активационный анализ был применен для определения бромидов в сигаретах и других материалах. Газохроматографические методы Для определения галогенидов можно использовать и газохроматографические методы: разработан быстрый метод разделения и определения малых концентраций галогенидов с использованием метода газовой хроматографии.[1] 1.3 Методы определения иодид-ионов
Спектрофотометрическое определение иодид-ионов Многие спектрофотометрические методы определения хлоридов и бромидов можно использовать и для определения иодидов. К ним относятся следующие методы. . Меркуриметрический роданидный метод. Сравнимый с ним метод, основанный на применении роданида серебра, также применим для определения бромида и иодида. . Метод с применением комплексов ртути с дифенилкарбазоном и дифенилкарбазидом. . Метод, основанный на вытеснении галогенидом HCN из цианида ртути. . Метод с использованием комплексоната ртути(II) (для бромида). . Метод, основанный на образовании галогенидов фенилртути и их взаимодействии с диэтилдитиокарбаминатом натрия. . Методы с использованием трифенилметановых красителей (для бромида).[1] В работе [18] изучены реакции взаимодействия иодида с индикаторами сульфофталеинового ряда. При этом предложен простой и достаточно селективный метод определения I-, основанный на их окислении иодатом калия в кислой среде с последующим взаимодействием выделившегося иода с иодфеноловым красным. Также применим чувствительный колориметрический метод микро определения йода в морской воде, который предложен в работе [19]. В источнике [20] описано исследование простой и точной методики спектрофотометрического определения I-, основанной на его каталитическом эффекте на окисление 4, 4-бис-(диметиламино)дифенилметана с помощью хлорамина Т в слабокислой среде. Аргентометрические методы Иодиды можно определять методом Фольгарда. Титрование по Мору непригодно для определения иодида, так как хромат сорбируется осадком, что затрудняет индикацию точки эквивалентности.[1] Для титрования иодидов можно использовать метод адсорбционных индикаторов. Иодид нельзя титровать в присутствии хлорида и бромида, так как эти ионы сорбируются поверхностью осадка, что приводит к завышению результатов определения иодидов.[1] Редокс-методы Иодид можно определяются окислением его до 1ода, I+ или иодата. Этот метод обладает тем преимуществом, что бромид и хлорид не мешают определению, если их содержание не очень велико. Эмиссионный спектральный анализ. Для иодида характерно образование интенсивных молекулярных эмиссионных полос в присутствии индия в холодном водородно-азотном диффузионном пламени. Это используется в эмиссионном спектральном анализе I-ионов. Флуориметрический метод Для флуориметрического определения 20 мкг иодида используют ацетат уранила. Флуоресценцию измеряют при 520 нм, длина возбуждающего излучения света 365 нм. Атомно-абсорбционный метод В атомно-абсорбционном анализе Киркбрайт, Вест и Вильсон определяли иодид по резонансной линии 183, 0 нм в пламени оксид азота (I) - ацетилен. Предел определения - 12 ррm для иодата, иодида или периодата. Разработан косвенный ААС-метод определения иодида, сравнимый по характеристикам с выше упомянутым методом. Потенциометрические методы, ионоселективные электроды Методы аргентометрического и меркуриметрического потенциометрического титрования иодида аналогичны таковым для определения бромида. Существуют методики, по которым можно определять микро концентрации иодидов на фоне 2Ч105-кратного избытка хлорида методом потенциометрии до нулевой точки. При этом применяют электроды серебро-иодид серебра и платина. Существует йодидный ионоселективный электрод, который характеризуется лучшей селективностью по сравнению с другими анионоселективными электродами. Определению иодида не мешают 1000-кратные избытки концентрации фторида, хлорида и бромида, а цианид и сульфид мешают определению. Кулонометрические методы Иодиды, так же как бромиды и хлориды, можно определять кулонометрическим методом. Чаще всего их титруют электрогенерированным ионом серебра. Для титрования микроконцетраций иодида используют электрогенерированный бром, который окисляет иодид до иодата. Ошибка определения 50 мкг иодида меньше 1%. Хлорид не мешает определению. Для определения малых количеств иодид-иона (0, 1-1, 3 мкг) применяют метод кулонометрии [21] при постоянном потенциале (+0, 75 В относительно насыщенного каломельного электрода) на фоне 0, 1 н серной кислоты в ячейке специальной конструкции, использующей Pt-электрод большой поверхности (400 см2) и принудительную циркуляцию анализируемого электролита с помощью центробежного стеклянного насоса. Точность и воспроизводимость определения I- лежит в пределах ± 0, 1%. Окисление I--ионов на фоне 0, 1 н H2SO4 протекает до образования I2; в присутствии FeCl3 (~ 0, 1М) образуется ICl2-. При потенциале +0, 95 В, однако и в этом случае окисление идет количественно и ошибка определения йодида не превышает 2% (а при содержании в пробе ~ 1мкг иодида ошибка составляет ~0, 5%). Амперометрические методы Известно, что Милуанова и Сонгина разработали амперометрический метод определения иодида с использованием К2Сr2О7. Титрование проводят в 2 М H2S04. He мешают определению 1000-кратный избыток хлорида и 2000-кратный избыток бромида. Относительная ошибка определения 2-3 мг иодида ниже 0, 4%.[1] Кинетические методы Известен кинетический метод определения 0, 01 - 1 мкг иодида, основанный на его каталитическом действии на реакцию церий(IV) - мышьяк(III). Каталиметрические методы способны обеспечить определение ультра малых его концентраций в присутствии различных ионов. Высокая чувствительность метода позволяет проводить непосредственное определение иодида, минуя длительную стадию его предварительного концентрирования. ОБОСНОВАНИЕ ТЕМЫ РАБОТЫ
Работа поставлена с целью определения методом потенциостатической кулонометрии бромид- и йодид-ионов при их совместном присутствии. Задача поэлементного определения галогенид-ионов в смеси до настоящего времени разрешима только методом ионной хроматографии. В анализе галогенид - содержащих объектов разработка альтернативных методик, основанных на разных принципах, является актуальной задачей.. Определение с использованием альтернативных методик позволяет обосновать правильность результатов анализа. Исследование посвящено изучению вариантов определения бромид- и йодид-ионов при их совместном присутствии методом потенциостатической кулонометрии на платиновых электродах. Вариант предполагает сопоставление зависимости времени выхода по току от концентрации бромид- и йодид-ионов и определение бромидов и йодидов из параллельных проб (без отделения йода) при выбранном потенциале. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
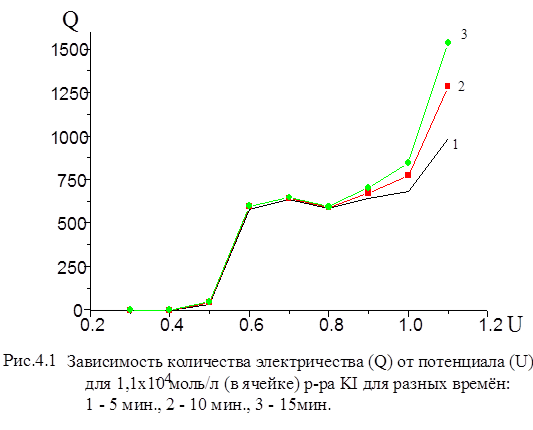
Метод потенциостатической кулонометрии относится к группе электрохимических методов и является абсолютным. Он позволяет судить о содержании анализируемого вещества в растворе путем измерения количества электричества, необходимого для полного превращения анализируемого вещества в ходе его электролитического восстановления (окисления) при условии 100 %-ного выхода по току (эффективности тока генерации). Если электролиз проводят в условиях, когда смешение катодных и анодных продуктов исключено, то все количество электричества, прошедшее через раствор в процессе электролиза, расходуется только на окисление (анодная реакция) или восстановление (катодная реакция) единственного вещества. Количество электричества, израсходованного за время протекания реакции до полного разложения реагирующего вещества, определяют содержание этого вещества, основываясь на известных законах электролиза. Единица количества электричества - кулон (Кл, А-с)' и дала название методу анализа -кулонометрия.[28] Соотношение между количеством электричества и массой превратившегося в ходе электролиза вещества установлено законом Фарадея:
т = Mit/nF,
где т - масса прореагировавшего (определяемого) вещества, г; М - его молярная масса, г/моль; i - сила тока в цепи, А; т - время электролиза, с; п - число электронов, принимающих участие в реакции с 1 молекулой определяемого вещества; F-постоянная Фарадея, равная 96500 Кл/моль (А-с/моль). Значение потенциала рабочего электрода, которое надо задать для определения интересующего вещества, можно установить разными способами. Электродный потенциал Е для любой концентрации электродноактивных веществ можно вычислить с помощью уравнения Нернста, записанного в форме:
E Ox/Red = E0* + RT/(nF)ln(COx/CRed) 0* - формальный потенциал оксред системы. На практике для проведения электролиза потенциал рабочего электрода устанавливают более отрицательным (при восстановлении) или более положительным (при окислении) относительно формального значения потенциала. Величина сдвига Е определяется желаемой степенью завершенности электрохимического превращения вещества. В этом случае сила тока, протекающего при электролизе в стационарных условиях, определяется уравнением:
I = I д. пр.[1-exp (nF/(RT)DE)] д. пр -сила предельного тока, который может быть получен при данной концентрации вещества в случае диффузионного контроля скорости переноса вещества к электроду. Можно вести электролиз на предельной силе. Поскольку сила предельного тока пропорциональна концентрации, то по мере протекания электролиза регистрируемая сила тока будет падать (Е = const по условию) в соответствии с убылью концентрации вещества в растворе. Падение силы тока описывается уравнением:
I=I exp(-kt)
I -сила тока в начальный момент времени электролиза, А; k - константа; t время, прошедшее от начала электролиза до момента измерения тока, с. Последнее уравнение позволяет вычислить время электролиза, необходимое для разложения анализируемого вещества с любой полнотой, если известна константа k. Значение k зависит от коэффициента диффузии электроактивного вещества D, площади поверхности рабочего электрода А, объёма анализируемого раствора V и толщины диффузионного слоя по уравнению =DA/(V .
В полулогарифмических координатах (lgI - t ) эта зависимость имеет вид прямой линии. Экспериментальная кривая зависимости, I от имеет более сложный вид. Искажения на начальном участке связаны с токами заряжения двойного электрического слоя (ДЭС), на конечном - с влиянием разложения неизбежных примесей. Имеются и другие причины нарушения хода кривой.[28] Инструментальные погрешности кулонометрической установки с контролируемым потенциалом можно разделить на две основные группы. Погрешности I группы - погрешности коэффициента передачи процесса “масса определяемого вещества ® отсчет прибора”. Основными составляющими погрешности являются: 22.Величина выхода по току электролитической ячейки (отличие от 100%). Эта составляющая полностью определяется конструкцией ячейки (отсутствие потерь вещества на стенках и диафрагмах, устранение брызг и “карманов” и т.п.) и может быть сведена к величине менее 0, 01%; 23.Коэффициент завершенности процесса электролиза. Погрешность величины коэффициента завершенности определяется факторами: · погрешность установки потенциала рабочего электрода. При потенциалах, отличающихся от формального на ±180 мВ, неточность установки потенциала в 1мВ изменяет величину коэффициента завершенности на 0, 004%; · нестабильность поддержания потенциала вследствие пульсаций тока электролиза и температурной нестабильности потенциала в процессе анализа. При оптимальной конструкции потенциостата этот вид погрешности может быть снижен до 0, 001-0, 003%; · изменение степени завершенности процесса электролиза при изменении постоянной времени ячейки из-за изменения величины не учитываемой части электролизного тока. При соотношении продолжительности электролиза T и постоянной времени ячейки τ, равном T/τ =12 (T=600c, τ =50c), изменение постоянной времени на 15% приводит к дополнительной погрешности, равной 0, 003%, а изменение на 30% приводит к погрешности 0, 02%. 1. Нелинейность интегратора тока электролиза изменяет общую величину интеграла. В большинстве интеграторов нелинейность увеличивается обратно пропорционально величине интегрируемого сигнала. Особой разновидностью нелинейности является погрешность дискретности цифровых интеграторов, которая также растет обратно пропорционально величине тока и должна обязательно учитываться. 2. Коэффициент передачи интегратора (цена единицы отсчета)- прямо влияет на погрешность кулонометрического анализа. Составляющими этой погрешности являются: · погрешность, определяемая временной нестабильностью коэффициента передачи интегратора. Данная составляющая может быть существенно снижена периодической градуировкой; · погрешность вследствие изменения коэффициента передачи интегратора при изменении температуры. Эта погрешность, в основном, определяется стабильностью измерительного резистора. Суммарная величина этой погрешности может быть доведена до 0, 0003-0, 0006% на 1°С и может быть дополнительно снижена термостатированием помещения и отдельных узлов кулонометрической установки. Погрешности II группы- погрешности градуировки кулонометрической установки. К ним можно отнести: 1. Погрешности градуировки задатчика потенциала. Практически полностью определяется погрешностью вольтметра постоянного тока, используемого при градуировке, и числом разрядов цифроаналогового преобразователя, синтезирующего выходное напряжение задатчика. При использовании лучших моделей вольтметров погрешность градуировки не превышает 0, 1-0, 2 мВ. 2. Погрешность градуировки интегратора (определение цены единицы отсчета). Определяется следующими факторами: · погрешность задания эталонного тока (тока градуировки). Определяется погрешностями вольтметра постоянного тока и катушки образцового сопротивления, по которым вычисляется величина тока. Реально эта величина может быть снижена до 0, 01%. Суммарная величина инструментальной погрешности может быть доведена до 0, 02% (при изменении температуры на 1°С) или 0, 04% (при изменении температуры на 10°С). При этом определяющими факторами являются величина выхода по току электролитической ячейки, нелинейность интегратора тока электролиза и погрешность градуировки интегратора (цены единицы отсчета). На рис. 4.1 приведена измеренная нами зависимость количества электричества от потенциала рабочего электрода для иодид-иона, из которой видно, что в области потенциалов U = 0, 6 - 0, 8 В выход йода по току в течении 10 - 15 мин приближается к 99 ±1%.
Точное значение мы не определяли, но из градуировочной зависимости количества электричества от концентрации иодид-ионов, при решении обратной задачи-нахождение концентрации по величине Q - определили, что погрешность не превышает 1%. На рис. 4.2 приведена аналогичная зависимость для бромид - иона. В области потенциалов 1, 2 -1, 3 В наблюдается “плато”.
Рис. 4.2 Зависимость количества электричества (Q) от потенциала (U) для 1, 1´ 10-4 моль/л раствора KBr (в ячейке).
При измерении временных зависимостей при U = 1, 2 В Q при фиксированной концентрации бромид-иона проявляется существенное уменьшение скорости электрохимической реакции (рис.4.3, нижняя кривая). Попытка измерить градуировочную зависимость Q - C при U = 1, 2 В удовлетворительных результатов не дала. По нашему мнению, это связано с возможным частичным окислением Pt на рабочем электроде В связи с выше сказанным мы были вынуждены уменьшить рабочий потенциал для выделения брома до U = 1, 1 В. При измерении количества электричества для бромид-иона было замечено, что в присутствии следовых количеств иодид-ионанаблюдается ускорение электрохимического окисления бромида до брома (рис. 3, верхняя кривая).
Рис. 4.3 Окисление бромид-ионов с добавкой и без добавки иодид-ионов при разных потенциалах
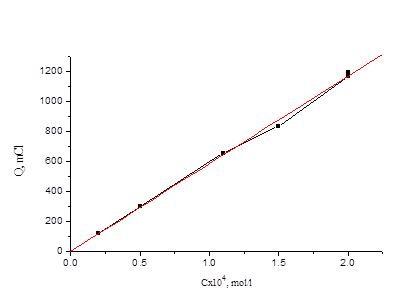
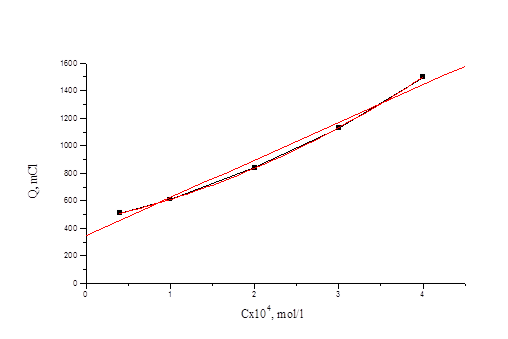
Из рис. 4.1 видно, что при потенциале U = 1, 1 В для иодида характерно более глубокое окисление. Скорость реакции окисления иодида достаточно высокая, а продукты могут взаимодействовать с бромид-ионами, что в результате и ускоряет реакцию окисления бромид-иона. На рис. 3 (верхняя кривая) - зависимость Q от времени для бромид-ионов в присутствии иодид-ионов с концентрацией, соответствующей 0, 1 % от концентрации бромида. Кулонометрическое определение модельной смеси I и Br заключается в следующем: раствор, содержащий аликвоты растворов бромид-и иодид-ионов помещаем в ячейку и ведем электролиз при потенциале U = +0, 75 В в течение 15 минут. Затем данный раствор помещаем в делительную воронку и четырехкратным экстрагированием четыреххлористым углеродом извлекаем выделившийся йод. Аликвоту оставшегося раствора переносим в мерную колбу, разбавляем и проводим электролиз данного раствора при потенциале U = 1, 1 В. По количеству выделившегося электричества рассчитываем концентрации иодид-и бромид-ионов. На рис. 4.4, 4.5 приведены градуировочные зависимости (Q от с) для иодид-иона и для бромид-иона в присутствии небольших (“остаточных”) количеств иодида.
Рис. 4.4 Зависимость количества электричества от концентрации иодид-иона для растворов смеси иодид- и бромид-ионов.
Рис. 4.5 Зависимость количества электричества от концентрации бромид-иона для растворов смеси иодид- и бромид-ионов.
Как видно, строгая линейная зависимость выполняется только для иодида, а для бромида зависимость можно апроксимировать линией. При числе градуировочных растворов-5 в исследованном нами диапазоне концентраций можно использовать и нелинейный градуировочный график. Продолжая исследования, в первую очередь, были получены зависимости количества электричества от времени для йодид-ионов при потенциале выделения брома (U=1, 1 В). При данном потенциале для йодида характерно более глубокое окисление предположительно до йодата. Исходя из этого, мы рассчитывали, какое количества электричества при этом должно выделится. Из рис. 4 видно, что при потенциале 1, 1 В теоретический выход при больших концентрациях (порядка 10-4 моль/л) достигается за длительный промежуток времени, т.е. электрохимическое окисление идет медленно. |
Последнее изменение этой страницы: 2020-02-17; Просмотров: 157; Нарушение авторского права страницы