
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Предмет «физическая и коллоидная химия».Стр 1 из 8Следующая ⇒
Предмет «физическая и коллоидная химия». Предметом физической химии является объяснение химических явлений на основе более общих законов физики. Физическая химия рассматривает две основные группы вопросов: 1. Изучение строения и свойств вещества и составляющих его частиц; 2. Изучение процессов взаимодействия веществ. В курсе физической химии обычно выделяют несколько разделов. Строение вещества. В этот раздел входят учение о строении атомов и молекул и учение об агрегатных состояниях вещества. Изучение строение вещества необходимо для выяснения важнейших вопросов об образовании молекул из атомов, о природе химической связи, о строении и взаимодействии молекул. Именно в этой своей части физическая химия очень тесно переплетается со всеми направлениями современной химии, поскольку изучение химических свойств вещества вне связи со строением атомов и молекул на современном уровне невозможно. Химическая термодинамика изучает энергетические эффекты химических процессов; позволяет определить возможность, направление и глубину протекания химического процесса в конкретных условиях. Химическая кинетика. В этом разделе физической химии изучается скорость и механизм протекания химических процессов в различных средах при различных условиях. Учение о растворах рассматривает процессы образования растворов, их внутреннюю структуру и важнейшие свойства, зависимость структуры и свойств от природы компонентов раствора. Электрохимия изучает особенности свойств растворов электролитов, явления электропроводности, электролиза, коррозии, работу гальванических элементов. Коллоидная химия изучает поверхностные явления и свойства мелкодисперсных гетерогенных систем.Все разделы физической химии объединяет единая основа – общие законы природы, которые применимы к любым процессам и любым системам, независимо от их строения.
Что изучает химическая термодинамика. Термодинамика – наука о взаимопревращениях различных форм энергии и законах этих превращений. Термодинамика базируется только на экспериментально обнаруженных объективных закономерностях, выраженных в двух основных началах термодинамики. Термодинамика изучает: 1.Переходы энергии из одной формы в другую, от одной части системы к другой; 2.Энергетические эффекты, сопровождающие различные физические и химические процессы и зависимость их от условий протекания данных процессов; 3.Возможность, направление и пределы самопроизвольного протекания процессов в рассматриваемых условиях. Необходимо отметить, что классическая термодинамика имеет следующие ограничения: 1.Термодинамика не рассматривает внутреннее строение тел и механизм протекающих в них процессов; 2.Классическая термодинамика изучает только макроскопические системы; 3.В термодинамике отсутствует понятие " время".
Основные понятия термодинамики. Термодинамическая система – тело или группа тел, находящихся во взаимодействии, мысленно или реально обособленные от окружающей среды. Гомогенная система – система, внутри которой нет поверхностей, разделяющих отличающиеся по свойствам части системы (фазы). Гетерогенная система – система, внутри которой присутствуют поверхности, разделяющие отличающиеся по свойствам части системы. Фаза – совокупность гомогенных частей гетерогенной системы, одинаковых по физическим и химическим свойствам, отделённая от других частей системы видимыми поверхностями раздела. Изолированная система – система, которая не обменивается с окружающей средой ни веществом, ни энергией.Закрытая система – система, которая обменивается с окружающей средой энергией, но не обменивается веществом. Открытая система – система, которая обменивается с окружающей средой и веществом, и энергией.Совокупность всех физических и химических свойств системы характеризует её термодинамическое состояние. Все величины, характеризующие какое-либо макроскопическое свойство рассматриваемой системы – параметры состояния. Опытным путем установлено, что для однозначной характеристики данной системы необходимо использовать некоторое число параметров, называемых независимыми; все остальные параметры рассматриваются как функции независимых параметров. В качестве независимых параметров состояния обычно выбирают параметры, поддающиеся непосредственному измерению, например температуру, давление, концентрацию и т.д. Всякое изменение термодинамического состояния системы (изменения хотя бы одного параметра состояния) есть термодинамический процесс. Обратимый процесс – процесс, допускающий возможность возвращения системы в исходное состояние без того, чтобы в окружающей среде остались какие-либо изменения. Равновесный процесс – процесс, при котором система проходит через непрерывный ряд равновесных состояний. Энергия - мера способности системы совершать работу; общая качественная мера движения и взаимодействия материи. Энергия является неотъемлемым свойством материи. Различают потенциальную энергию, обусловленную положением тела в поле некоторых сил, и кинетическую энергию, обусловленную изменением положения тела в пространстве. Внутренняя энергия системы – сумма кинетической и потенциальной энергии всех частиц, составляющих систему. Можно также определить внутреннюю энергию системы как её полную энергию за вычетом кинетической и потенциальной энергии системы как целого.
4.Основные формулировки первого начала термодинамики. Первое начало термодинамики представляет собой закон сохранения энергии, один из всеобщих законов природы (наряду с законами сохранения импульса, заряда и симметрии): Энергия неуничтожаема и несотворяема; она может только переходить из одной формы в другую в эквивалентных соотношениях.Первое начало термодинамики представляет собой постулат – оно не может быть доказано логическим путем или выведено из каких-либо более общих положений. Истинность этого постулата подтверждается тем, что ни одно из его следствий не находится в противоречии с опытом. Приведем еще некоторые формулировки первого начала термодинамики: Полная энергия изолированной системы постоянна; Невозможен вечный двигатель первого рода (двигатель, совершающий работу без затраты энергии).Первое начало термодинамики устанавливает соотношение между теплотой Q, работой А и изменением внутренней энергии системы Δ U: Изменение внутренней энергии системы равно количеству сообщенной системе теплоты минус количество работы, совершенной системой против внешних сил. Внутренняя энергия является функцией состояния; это означает, что изменение внутренней энергии Δ U не зависит от пути перехода системы из состояния 1 в состояние 2 и равно разности величин внутренней энергии U2 и U1 в этих состояниях:
Следует отметить, что определить абсолютное значение внутренней энергии системы невозможно; термодинамику интересует лишь изменение внутренней энергии в ходе какого-либо процесса.
5.Применение первого начала термодинамики к различным процессам. Рассмотрим приложение первого начала термодинамики для определения работы, совершаемой системой при различных термодинамических процессах. Изохорный процесс (V = const; Δ V = 0). Поскольку работа расширения равна произведению давления и изменения объема, для изохорного процесса получаем:
Изотермический процесс (Т = const). Из уравнения состояния одного моля идеального газа получаем:
Проинтегрировав выражение (I.6) от V1 до V2, получим
Изобарный процесс (Р = const).
Подставляя полученные выражения для работы различных процессов в уравнение (I.1), для тепловых эффектов этих процессов получим:
В уравнении (I.12) сгруппируем переменные с одинаковыми индексами. Получаем:
Введем новую функцию состояния системы – энтальпию H, тождественно равную сумме внутренней энергии и произведения давления на объем:
Тогда выражение (I.13) преобразуется к следующему виду:
Т.о., тепловой эффект изобарного процесса равен изменению энтальпии системы. Адиабатический процесс (Q = 0). При адиабатическом процессе работа расширения совершается за счёт уменьшения внутренней энергии газа:
В случае если Cv не зависит от температуры (что справедливо для многих реальных газов), работа, произведённая газом при его адиабатическом расширении, прямо пропорциональна разности температур:
Закон Гесса. Тепловые эффекты, сопровождающие протекание химических реакций, являются предметом одного из разделов химической термодинамики – термохимии. Определим некоторые понятия термохимии. Теплота образования вещества – тепловой эффект реакции образования 1 моля сложного вещества из простых. Теплоты образования простых веществ принимаются равными нулю. Теплота сгорания вещества – тепловой эффект реакции окисления 1 моля вещества в избытке кислорода до высших устойчивых оксидов. Теплота растворения – тепловой эффект процесса растворения 1 моля вещества в бесконечно большом количестве растворителя. Теплота растворения складывается из двух составляющих: теплоты разрушения кристаллической решетки (для твердого вещества) и теплоты сольватации:
Поскольку Δ Нкр.реш всегда положительно (на разрушение кристаллической решетки необходимо затратить энергию), а Δ Нсольв всегда отрицательно, знак Δ Нраств определяется соотношением абсолютных величин Δ Нкр.реш. и Δ Нсольв:
Основным законом термохимии является закон Гесса, являющийся частным случаем первого начала термодинамики: Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания. Выше было показано, что изменение энтальпии Δ Н (тепловой эффект изобарного процесса Qp) и изменение внутренней энергии Δ U(тепловой эффект изохорного процесса Qv) не зависят от пути, по которому система переходит из начального состояния в конечное. Согласно закону Гесса, тепловые эффекты всех этих реакций связаны следующим соотношением:
Следствие из закона Гесса. Практическое значение закона Гесса состоит в том, что он позволяет рассчитывать тепловые эффекты химических процессов. В термохимических расчетах обычно используют ряд следствий из закона Гесса: 1. Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье – Лапласа).2. Для двух реакций, имеющих одинаковые исходные, но разные конечные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного конечного состояния в другое. С + О2 ––> СО + 1/2 О2 Δ Н 1 С + О2 ––> СО2 Δ Н 2 СО + 1/2 О2 ––> СО2 Δ Н 3
3. Для двух реакций, имеющих одинаковые конечные, но разные исходные состояния, разность тепловых эффектов представляет собой тепловой эффект перехода из одного исходного состояния в другое. С(алмаз) + О2 ––> СО2 Δ Н 1 С(графит) + О2 ––> СО2 Δ Н 2 С(алмаз) ––> С(графит) Δ Н 3
4. Тепловой эффект химической реакции равен разности сумм теплот образования продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты.
5. Тепловой эффект химической реакции равен разности сумм теплот сгорания исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты.
8. Зависимость теплового эффекта реакции от температуры. Закон Кирхгоффа В общем случае тепловой эффект химической реакции зависит от температуры и давления, при которых проводится реакция. Влиянием давления на Δ Н и Δ U реакции обычно пренебрегают. Влияние температуры на величины тепловых эффектов описывает закон Кирхгоффа: Температурный коэффициент теплового эффекта химической реакции равен изменению теплоемкости системы в ходе реакции.Продифференцируем Δ Н и Δ U по температуре при постоянных давлении и температуре соответственно:
Производные энтальпии и внутренней энергии системы по температуре есть теплоемкости системы в изобарных и изохорных условиях Cp и Cv соответственно:
Подставив выражения (I.24, I.25) в (I.22, I.23), получаем математическую запись закона Кирхгоффа:
Для химического процесса изменение теплоемкости задается изменением состава системы и рассчитывается следующим образом:
Энтропия. Величина связанной энергии тем больше, чем меньше разность температур, и при T = const тепловая машина не может производить работу. Мерой связанной энергии является новая термодинамическая функция состояния, называемая энтропией. Введем определение энтропии, основываясь на цикле Карно. Преобразуем выражение (I.41) к следующему виду:
Отсюда получаем, что для обратимого цикла Карно отношение количества теплоты к температуре, при которой теплота передана системе (т.н. приведенная теплота) есть величина постоянная:
Это верно для любого обратимого циклического процесса, т.к. его можно представить в виде суммы элементарных циклов Карно, для каждого из которых
Т.о., алгебраическая сумма приведённых теплот для произвольного обратимого цикла равна нулю:
Выражение (I.49) для любого цикла может быть заменено интегралом по замкнутому контуру:
Если интеграл по замкнутому контуру равен нулю, то подынтегральное выражение есть полный дифференциал некоторой функции состояния; эта функция состояния есть энтропия S: Выражение (I.51) является определением новой функции состояния – энтропии и математической записью второго начала термодинамики для обратимых процессов. Если система обратимо переходит из состояния 1 в состояние 2, изменение энтропии будет равно:
Подставляя (I.51, I.52) в выражения для первого начала термодинамики (I.1, I.2) получим совместное аналитическое выражение двух начал термодинамики для обратимых процессов:
Сильные электролиты Предположение Аррениуса о том, что в растворе сильного электролита также существует динамическое равновесие между молекулами и ионами, как и у слабых электролитов, оказалось ошибочным. Экспериментальные исследования показали, что, во-первых, величина константы диссоциации сильного электролита зависит от концентрации (т.е. к растворам сильных электролитов неприменим закон действующих масс) и, во-вторых, никакими методами не удалось обнаружить в растворах сильных электролитов непродиссоциировавшие молекулы. Это позволило сделать вывод, что сильные электролиты в растворах любых концентраций полностью диссоциируют на ионы и, следовательно, закономерности, полученные для слабых электролитов, не могут применяться к сильным электролитам без соответствующих поправок. Качественная теория сильных электролитов была разработана П.Дебаем и Г.Хюккелем (1923). Чем выше концентрация раствора, тем сильнее взаимодействие ионов, тем меньше и кажущаяся степень диссоциации сильного электролита. Количественные расчеты характеристик растворов сильных электролитов осуществляют с помощью понятий активности электролита а э и активностей катионов и анионов а + и а - соответственно, которые равны произведению коэффициента активности на концентрацию:
Для бинарного электролита средняя активность электролита связана с активностями ионов соотношением (III.28); подобным же образом связан средний коэффициент активности с ионными:
Теория Дебая – Хюккеля применима только при концентрациях, не превышающих 0.05 моль/л. Для более концентрированных растворов сильных электролитов количественной теории не существует. Классификация электродов По типу электродной реакции все электроды можно разделить на две группы Электроды первого рода К электродам первого рода относятся электроды, состоящие из металлической пластинки, погруженной в раствор соли того же металла. При обратимой работе элемента, в который включен электрод, на металлической пластинке идет процесс перехода катионов из металла в раствор либо из раствора в металл. Т.о., электроды первого рода обратимы по катиону и их потенциал связан уравнением Нернста (1) с концентрацией катиона
Электроды второго рода Электродами второго рода являются электроды, в которых металл покрыт малорастворимой солью этого металла и находится в растворе, содержащем другую растворимую соль с тем же анионом. Электроды этого типа обратимы относительно аниона и зависимость их электродного потенциала от температуры и концентрации аниона может быть записана в следующем виде:
Электроды сравнения Для определения электродного потенциала элемента необходимо измерить ЭДС гальванического элемента, составленного из испытуемого электрода и электрода с точно известным потенциалом – электрода сравнения. В качестве примеров рассмотрим водородный, каломельный и хлорсеребряный электроды. Водородный электрод представляет собой платиновую пластинку, омываемую газообразным водородом, погруженную в раствор, содержащий ионы водорода. Потенциал водородного электрода зависит от активности ионов Н+ в растворе и давления водорода; потенциал стандартного водородного электрода (с активностью ионов Н+ 1 моль/л и давлением водорода 101.3 кПа) принят равным нулю. Поэтому для электродного потенциала нестандартного водородного электрода можно записать: Каломельный электрод. Работа с водородным электродом довольно неудобна, поэтому в качестве электрода сравнения часто используется более простой в обращении каломельный электрод, величина электродного потенциала которого относительно стандартного водородного электрода точно известна и зависит только от температуры. Каломельный электрод состоит из ртутного электрода, помещенного в раствор КСl определенной концентрации и насыщенный каломелью Hg2Сl2: Нg / Нg2Сl2, КСl Каломельный электрод обратим относительно анионов хлора и уравнение Нернста для него имеет вид:
Хлорсеребряный электрод. В качестве электрода сравнения используют также другой электрод второго рода – хлорсеребряный, который также обратим относительно анионов хлора: Аg / АgСl, КСl Величина потенциала хлорсеребряного электрода зависит от активности ионов хлора; данная зависимость имеет следующий вид: Индикаторные электроды. В качестве индикаторного электрода может использоваться водородный электрод, однако работа с ним неудобна и на практике чаще применяются хингидронный и стеклянный электроды. Хингидронный электрод, относящийся к классу окислительно-восстановительных электродов представляет собой платиновую проволоку, опущенную в сосуд с исследуемым раствором, в который предварительно помещают избыточное количество хингидрона С6Н4О2·С6Н4(ОН)2 – соединения хинона С6Н4О2 и гидрохинона С6Н4(ОН)2, способных к взаимопревращению в равновесном окислительно-восстановительном процессе, в котором участвуют ионы водорода: С6Н4О2 + 2Н+ + 2е- ––> С6Н4(ОН)2 Хингидронный электрод является т.н. окислительно-восстановительным электродом; зависимость его потенциала от активности ионов водорода имеет следующий вид:
Стеклянный электрод, являющийся наиболее распространенным индикаторным электродом, относится к т.н. ионоселективным или мембранным электродам. Потенциал стеклянного электрода с водородной функцией выражается уравнением
Необходимо отметить, что стандартный потенциал ε oст для каждого электрода имеет свою величину, которая со временем изменяется; поэтому стеклянный электрод перед каждым измерением рН калибруется по стандартным буферным растворам с точно известным рН.
Скорость химической реакции Скорость химической реакции есть число элементарных актов химической реакции, происходящих в единицу времени в единице объема (для гомогенных реакций) или на единице поверхности (для гетерогенных реакций).Скорость химической реакции есть изменение концентрации реагирующих веществ в единицу времени. Наиболее часто в химии рассматривается зависимость концентрации реагентов от времени. В случае односторонних химических реакций очевидно, что концентрации исходных веществ во времени постоянно уменьшаются (Δ Сисх < 0), а концентрации продуктов реакции увеличиваются (Δ Спрод > 0). Скорость реакции считается положительной, поэтому математически определение средней скорости реакции в интервале времени Δ t записывается следующим образом:
В различных интервалах времени средняя скорость химической реакции имеет разные значения; истинная (мгновенная) скорость реакции определяется как производная от концентрации по времени:
Истинную скорость реакции можно определить графически, проведя касательную к кинетической кривой (рис.1); истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной:
Рис. 1 Графическое определение Vист.
Скорость химической реакции зависит от множества факторов: природы реагирующих веществ, их концентрации, температуры, природы растворителя и т.д. Реакции первого порядка Реакции первого порядка характеризуются кинетическим уравнением вида. Подставим в него выражение:
После интегрирования выражения (2) получаем:
Константу интегрирования g определим из начальных условий: в момент времени t = 0 концентрация С равна начальной концентрации Со. Отсюда следует, что g = ln Со. Получаем:
Т.о., логарифм концентрации для реакции первого порядка линейно зависит от времени (рис.1) и константа скорости численно равна тангенсу угла наклона прямой к оси времени.
Из уравнения (4) легко получить выражение для константы скорости односторонней реакции первого порядка:
Еще одной кинетической характеристикой реакции является период полупревращения t1/2 – время, за которое концентрация исходного вещества уменьшается вдвое по сравнению с исходной. Выразим t1/2 для реакции первого порядка, учитывая, что С = ½ Со:
Отсюда Как видно из полученного выражения, период полупревращения реакции первого порядка не зависит от начальной концентрации исходного вещества. Реакции второго порядка Для реакций второго порядка кинетическое уравнение имеет следующий вид:
либо Рассмотрим простейший случай, когда кинетическое уравнение имеет вид (1) или, что то же самое, в уравнении вида (2) концентрации исходных веществ одинаковы; уравнение (1) в этом случае можно переписать следующим образом:
Если начальные концентрации реагирующих веществ Cо, А и Cо, В различны, то константу скорости реакции находят интегрированием уравнения (8),
Порядок химической реакции есть формально-кинетическое понятие, физический смысл которого для элементарных (одностадийных) реакций заключается в следующем: порядок реакции равен числу одновременно изменяющихся концентраций. В случае элементарных реакций порядок реакции может быть равен сумме коэффициентов в стехиометрическом уравнении реакции; однако в общем случае порядок реакции определяется только из экспериментальных данных и зависит от условий проведения реакции. Уравнение Аррениуса Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. Аррениус постулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации. Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию. Рассмотрим путь некоторой элементарной реакции А + В ––> С Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения, называемого активированным комплексом: А ––> K# ––> B Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции. Рассмотрим термодинамический вывод выражения, описывающего зависимость константы скорости реакции от температуры и величины энергии активации – уравнения Аррениуса. Согласно уравнению изобары Вант-Гоффа,
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
Представив изменение энтальпии реакции Δ Hº в виде разности двух величин E1 и E2, получаем:
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации:
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
КАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ Скорость химической реакции при данной температуре определяется скоростью образования активированного комплекса, которая, в свою очередь, зависит от величины энергии активации. Во многих химических реакциях в структуру активированного комплекса могут входить вещества, стехиометрически не являющиеся реагентами. В случае наличия нескольких переходных состояний реакция будет идти в основном по пути с наименьшим активационным барьером. Катали з – явление изменения скорости химической реакции в присутствии веществ, состояние и количество которых после реакции остаются неизменными. Различают положительный и отрицательный катализ, хотя часто под термином " катализ" подразумевают только положительный катализ; отрицательный катализ называют ингибированием.Вещество, входящее в структуру активированного комплекса, но стехиометрически не являющееся реагентом, называется катализатором. Для всех катализаторов характерны такие общие свойства, как специфичность и селективность действия. Специфичность катализатора заключается в его способности ускорять только одну реакцию или группу однотипных реакций и не влиять на скорость других реакций Селективность катализатора – способность ускорять одну из возможных при данных условиях параллельных реакций. Причиной увеличения скорости реакции при положительном катализе является уменьшение энергии активации при протекании реакции через активированный комплекс с участием катализатора.Поскольку, согласно уравнению Аррениуса, константа скорости химической реакции находится в экспоненциальной зависимости от величины энергии активации, уменьшение последней вызывает значительное увеличение константы скорости. Для отношения констант скорости можно записать:
Если Δ EA = –50 кДж/моль, то отношение констант скоростей составит 27·105 раз. Необходимо отметить, что наличие катализатора не влияет на величину изменения термодинамического потенциала в результате процесса и, следовательно, никакой катализатор не может сделать возможным самопроизвольное протекание термодинамически невозможного процесса. Катализатор не изменяет величину константы равновесия для обратимых реакций; влияние катализатора в этом случае заключается только в ускорении достижения равновесного состояния.В зависимости от фазового состояния реагентов и катализатора различают гомогенный и гетерогенный катализ. Гомогенный катализ. Гомогенный катализ – каталитические реакции, в которых реагенты и катализатор находятся в одной фазе. В случае гомогенно-каталитических процессов катализатор образует с реагентами промежуточные реакционноспособные продукты. Рассмотрим некоторую реакцию А + В ––> С |
Последнее изменение этой страницы: 2017-03-17; Просмотров: 2772; Нарушение авторского права страницы
 (1)
(1)  (2) Уравнение (I.1) является математической записью 1-го начала термодинамики для конечного, уравнение (I.2) – для бесконечно малого изменения состояния системы.
(2) Уравнение (I.1) является математической записью 1-го начала термодинамики для конечного, уравнение (I.2) – для бесконечно малого изменения состояния системы. (3)
(3)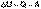 (I.1)
(I.1) (I.4)
(I.4)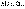 (I.5)
(I.5) (I.6)Отсюда:
(I.6)Отсюда:  (I.7)
(I.7) (I.8)
(I.8)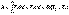 (I.9)
(I.9) (I.10)
(I.10) (I.11)
(I.11) (I.12)
(I.12) (I.13)
(I.13)
 (I.14)
(I.14) (I.15)
(I.15) (I.16)
(I.16)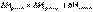

 .
. (I.18)
(I.18)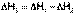 (I.19)
(I.19) (I.20)
(I.20) (I.21)
(I.21) (I.22)
(I.22) (I.23)
(I.23) (I.24)
(I.24) (I.25)
(I.25) (I.26)
(I.26) (I.27)
(I.27)

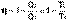 (I.45)
(I.45)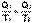 (I.46)
(I.46) 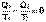 (I.47)
(I.47)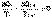 (I.48)
(I.48)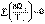 (I.49)
(I.49)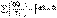 (I.50)
(I.50)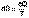 (I.51)
(I.51)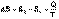 (I.52)
(I.52)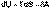
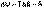 В изолированных системах самопроизвольно могут протекать только процессы, сопровождающиеся увеличением энтропии. Энтропия изолированной системы не может самопроизвольно убывать.Оба этих вывода также являются формулировками второго начала термодинамики.
В изолированных системах самопроизвольно могут протекать только процессы, сопровождающиеся увеличением энтропии. Энтропия изолированной системы не может самопроизвольно убывать.Оба этих вывода также являются формулировками второго начала термодинамики.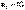 ;
; 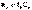
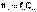
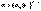
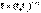 Дебаем и Хюккелем был разработан метод расчета среднего коэффициента активности сильного электролита. Для бинарного электролита уравнение имеет следующий вид:
Дебаем и Хюккелем был разработан метод расчета среднего коэффициента активности сильного электролита. Для бинарного электролита уравнение имеет следующий вид: 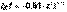 Здесь z – заряд иона, для которого рассчитывается коэффициент активности, I – т.н. ионная сила раствора: некоторый параметр, который одновременно учитывает молярную концентрацию и заряд всех имеющихся в растворе ионов. Ионная сила раствора равна полусумме концентраций всех ионов, умноженных на квадрат их заряда:
Здесь z – заряд иона, для которого рассчитывается коэффициент активности, I – т.н. ионная сила раствора: некоторый параметр, который одновременно учитывает молярную концентрацию и заряд всех имеющихся в растворе ионов. Ионная сила раствора равна полусумме концентраций всех ионов, умноженных на квадрат их заряда: 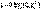
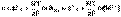 (1)
(1)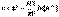 (2)
(2)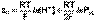 (3)
(3)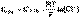 (4)
(4)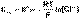 (5)
(5)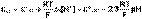 (6)
(6)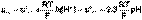 (7)
(7)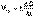 (1)
(1)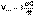 (2)
(2)
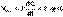 (3)
(3)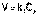 (1)
(1) 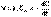 (2)
(2)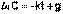 (3)
(3)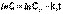 (4)
(4) Рис. 1 Зависимость логарифма концентрации от времени для реакций первого порядка.
Рис. 1 Зависимость логарифма концентрации от времени для реакций первого порядка.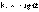 (5)
(5)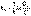 (6)
(6)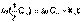 (7)
(7)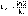 (8)
(8)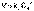 (1)
(1)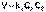 (2)
(2)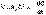 (3)После разделения переменных и интегрирования получаем:
(3)После разделения переменных и интегрирования получаем: 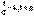 (4)Постоянную интегрирования g, как и в предыдущем случае, определим из начальных условий. Получим:
(4)Постоянную интегрирования g, как и в предыдущем случае, определим из начальных условий. Получим: 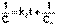 (5)Т.о., для реакций второго порядка, имеющих кинетическое уравнение вида (1), характерна линейная зависимость обратной концентрации от времени (рис.1) и константа скорости равна тангенсу угла наклона прямой к оси времени
(5)Т.о., для реакций второго порядка, имеющих кинетическое уравнение вида (1), характерна линейная зависимость обратной концентрации от времени (рис.1) и константа скорости равна тангенсу угла наклона прямой к оси времени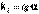 (6)
(6) 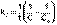 (7)
(7)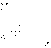 Рис. 2.4 Зависимость обратной концентрации от времени для реакций второго порядка.
Рис. 2.4 Зависимость обратной концентрации от времени для реакций второго порядка.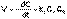 (8)В этом случае для константы скорости получаем выражение
(8)В этом случае для константы скорости получаем выражение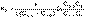 (9)
(9)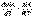 (II.31)
(II.31)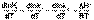 (II.32)
(II.32)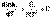 (II.33)
(II.33)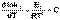 (II.34)
(II.34)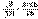 (II.35)
(II.35)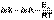 (II.36)
(II.36)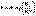 (II.37)
(II.37)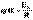 (II.38)
(II.38)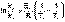 (II.39)
(II.39)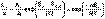 (II.44)
(II.44)