
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗАСтр 1 из 16Следующая ⇒
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Отчёт о лабораторной работе по дисциплине «Физико-химические методы анализа»
ЯГТУ 18.03.01 – 001 ЛР СОГЛАСОВАНО
Руководитель . хим. наук, доцент канд. экон. наук, доцент ____________С. А. Соловьева ____________М. А. Угрюмова
Отчёт выполнил студент группы ЗО-30 __________Ю.Б. Соловьёв 27.02.2017
2017
ФОТОМЕТРИЧЕСКИЙ МЕТОД АНАЛИЗА Теоретические основы
Фотометрический метод анализа основан на измерении интенсивности светового потока, прошедшего через анализируемый раствор. Основным законом фотометрии является закон Ламберта-Бугера-Бера (1.1): где Смысл закона Ламберта-Бугера-Бера можно выразить следующим образом. Одинаковые слои одного и того же вещества поглощают свет в одинаковой степени в независимости от интенсивности падающего на них светового потока. Если прологарифмировать уравнение (1.1) и изменить знаки на обратные, то уравнение принимает вид (1.2): где Т – светопропускание раствора, отн. ед. Величина Из уравнения (1.3) вытекает, что абсорбция раствора прямо пропорциональна концентрации анализируемого вещества и толщине слоя раствора. Существуют два метода фотометрического анализа:
1) Прямой фотометрический анализ. Метод основан на определении концентрации анализируемого вещества по закону Ламберта-Бугера-Бера (1.3). Аналитическим сигналом в этом методе является либо абсорбция, либо светопропускание растворов. Если вещество окрашено (e > 104 дм3/моль×см), то анализ проводят по методу собственного поглощения. В основном этот метод применяется для определения красителей или их смесей. В последнем случае при расчете концентраций отдельных компонентов смеси используют закон аддитивности абсорбций (1.4): где
Если вещество не окрашено (e = 0), то используют метод реагентов. Реагенты – это соединения, которые при взаимодействии с неокрашенным анализируемым веществом, образуют продукты реакции с высоким коэффициентом экстинкции. В методе реагентов используются реакции окисления, комплексообразования, синтеза красителей [2]. Абсорбция (светопропускание) анализируемых растворов измеряется на фотометрах или колориметрах. При измерении абсорбции используется относительный метод, который позволяет исключить поглощение света растворителем и учесть спектральные характеристики источников и приемников излучения. В качестве раствора сравнения используют либо чистый растворитель (метод «обычной», или «прямой» фотометрии), либо раствор анализируемого вещества с точно известной концентрацией (метод «дифференциальной» фотометрии). Метод обычной фотометрии используется для анализа растворов с абсорбцией до 0,8 отн. ед абсорбции. В методе дифференциальной фотометрии измеряется относительная абсорбция А отн = А ан. р-ра – А р-ра срав., что позволяет анализировать растворы с более высокой абсорбцией (более 0,8 отн.ед. абсорбции). 2) Косвенный метод – фотометрическое титрование. Фотометрическое титрование – это титриметрический метод анализа, в котором точка эквивалентности (Т.Э.) определяется по резкому изменению абсорбции в ходе титрования. С помощью этого метода можно изучать реакции, протекающие с изменением окраски растворов в ходе титрования. По результатам титрования вычерчивают кривую в координатах «абсорбция – объем титранта», аналитическим сигналом в этом методе является объем титранта в точке эквивалентности. В фотометрическом титровании используются реакции нейтрализации, осаждения, окислительно-восстановительные и комплексообразования. Метод позволяет проводить титрование по собственному поглощению и с помощью реагентов, в качестве последних могут использоваться индикаторы. Для фотометрического титрования по собственному поглощению необходимо, чтобы титруемые растворы или продукты реакции имели собственную характерную полосу поглощения в видимой области спектра. Если ни анализируемое вещество, ни титрант, ни продукты реакции не поглощают излучение в видимой области спектра, то применяют цветной индикатор. На практике титрование осуществляют либо в кювете с мешалкой при непрерывном добавлении титранта из бюретки к анализируемому раствору, либо титрант добавляется порциями в мерные колбы с одинаковой аликвотой анализируемого раствора, объем колб доводят растворителем до метки, перемешивают, а затем фотометрируют растворы. В первом случае удобно проводить титрование на монохроматоре, установив аналитическую длину волны в максимуме полосы поглощения окрашенного компонента или индикатора, во втором - на фотоколориметре. Фотометрический метод анализа используется для широкого диапазона определяемых концентраций от 10-3-10-4 до 20-30 % . Косвенный метод менее чувствительный, чем прямой метод, фотометрическое титрование, как правило, применяют для определения больших концентраций, порядка 10-2-10-3 моль/дм3. Селективность фотометрического метода довольно высокая, что позволяет анализировать сложные смеси веществ без их предварительного разделения. Погрешность фотометрического метода 0,1 –1,0 %.
Таблица 1.1 - Таблица дополнительных цветов
В последнем случае измеряют абсорбцию (согласно п.п. 4-7) стандартного раствора с наибольшей концентрацией со всеми имеющимися в приборе светофильтрами. Для работы выбирают светофильтр, при котором абсорбция стандартного раствора наибольшая. 4) Кюветы с анализируемым раствором и с раствором сравнения помещают в кюветное отделение. Кювету с раствором сравнения ставят в положение I, с анализируемым раствором – в положении II на панели фотоколориметра. 5) Поворотом ручки (положение I) вводят в световой поток кювету с чистым растворителем. С помощью ручки 100 % «грубо» и «точно» устанавливают стрелку прибора на ноль по нижней шкале (шкала абсорбции). Если стрелка не устанавливается на ноль, то следует подобрать другое положение ручки «чувствительность» (1, 2, 3). При измерениях со светофильтрами, помеченными на лицевой панели КФК-2 черным цветом, ручку «чувствительность» следует установить в положения 1-3, помеченные черным цветом. Аналогично поступают при измерениях со светофильтрами, помеченными красным цветом. 6) Поворотом ручки (положение II) вводят в световой поток кювету с анализируемым раствором. При этом крышка кюветного отделения должна быть закрыта. 7) Снимают отсчет по шкале абсорбции (нижняя шкала) с точностью до сотых, результат измерения заносят в таблицу. 8) По окончании работы прибор выключают, вынимают кюветы из кюветного отделения. Кюветы ополаскивают дистиллированной водой и протирают фильтровальной бумагой. Рабочее место приводят в порядок. Лабораторная работа № 1 Расчет результатов анализа
1) Расчет концентрации приготовленных стандартных растворов красителя
где Сисх.ст. р-ра – концентрация исходного стандартного раствора красителя в склянке, моль/дм3; Сст. – концентрация, приготовленного раствора в колбе, моль/дм3; Vисх.ст.р-ра – объем исходного стандартного раствора, отмеренный по бюретке, дм3; Vк – объем колбы, дм3. 2) Выбор светофильтра
Таблица 1 – Результаты фотометрирования стандартного раствора на разных светофильтрах
Заключение: для работы выбран светофильтр с lсв. = …, нм. 3) Результаты фотометрирования стандартных и анализируемого растворов на светофильтре с lсв. = …, нм
Таблица 2 – Результаты эксперимента
4) Построение калибровочного графика и определение концентрации красителя в анализируемом растворе. Калибровочный график строится по результатам, приведенным в таблице 2.

Результат проверяют у преподавателя или лаборанта, оформляют отчет по выполненной работе.
Вопросы к отчету по теме «Фотометрический метод анализа» 1 Фотометрический метод анализа. Законы поглощения света: закон Ламберта-Бугера-Бера, правило аддитивности абсорбции. Физический смысл коэффициента экстинкции и способы его определения. 2 Графическое изображение зависимости абсорбции от концентрации раствора. Причины, вызывающие отклонение от закона Ламберта-Бугера-Бера. Чувствительность фотометрического метода анализа. 3 Метод собственного поглощения и метод реагентов, область их применения. 4 Метод «обычной» и дифференциальной фотометрии. 5 Блок-схема фотоколориметра КФК-2, основные элементы прибора и их назначение. Относительный способ измерения абсорбции растворов. 6 Абсорбционные светофильтры, их характеристики и способы выбора.
Дополнительные вопросы
1 Особенности фотометрического анализа смеси красителей с непересекающимися и с пересекающимися спектрами. 2 Источники ультрафиолетового и видимого излучения, их устройство. 3 Фотонные приемники излучения: фотоэлементы с внешним фотоэффектом, фотоэлементы с внутренним фотоэффектом, фотоумножители. Требования к фотонным приемникам излучения, их устройство.
АБСОРБЦИОННАЯ СПЕКТРОСКОПИЯ Теоретические основы
Метод абсорбционной спектроскопии основан на изучении спектров поглощения анализируемых растворов в ультрафиолетовой, видимой и инфракрасной областях спектра. Спектры поглощения отражают вероятность поглощения анализируемым веществом электромагнитного излучения определенной длины волны и изображаются в координатах: А(l), Т(l), e(l), где А – абсорбция раствора, отн. ед.; Т – светопропускание, отн. ед.; e – молярный коэффициент ослабления (коэффициент экстинкции), дм3/моль×см; l – длина волны электромагнитного излучения, нм. Вместо длины волны может быть использована либо частота (n), либо волновое число ( В аналитической химии спектры поглощения используются для качественного, количественного анализа индивидуальных веществ и их смесей, определения термодинамических величин (констант диссоциации красителей, энтальпии и др.), структуры органических молекул, изучения механизма кинетических реакций и ряда других важнейших параметров. Качественный анализ в абсорбционной спектроскопии основан на специфичности спектров поглощения. Практически нет случаев, чтобы различные по химическому строению вещества имели бы совпадающие спектры поглощения в видимой, ультрафиолетовой и инфракрасной областях спектра. Министерства, ведомства и иностранные фирмы издают атласы спектров выпускаемых продуктов. В атласах содержатся спектры только тех веществ, технология или методика получения которых аттестована. Для идентификации исследуемого вещества записывают его спектр поглощения в определенном растворителе и сравнивают полученные данные с соответствующим спектром в атласе. Количественный анализ индивидуальных веществ основан на зависимости абсорбции растворов от концентрации согласно закону Ламберта-Бугера-Бера: А = eС×l [3]. Методы определения концентраций в абсорбционной спектроскопии аналогичны методам в фотометрии (метод молекулярного коэффициента экстинкции, метод двух растворов и др. [2]). Абсорбция измеряется на аналитической длине волны, которая выбирается, как правило, в максимуме полосы поглощения анализируемого вещества. Количественный анализ смеси невзаимодействующих веществ, спектры которых пересекают друг друга, требует использование закона аддитивности абсорбцией Основными преимуществами метода абсорбционной спектроскопии являются быстрота проведения анализа, широкая область применения, возможность анализа сложных смесей без их предварительного разделения, высокая чувствительность. Погрешность метода составляет 0,2-1,0 %.
Рисунок 3.5 – Блок-схема двулучевого спектрофотометра СФ-10: 1 - источник полихроматического света; 2 – монохроматор; 3, 3' - кюветы с раствором сравнения и анализируемым раствором, соответственно; 4, 4' - приемник излучения; 5 - схема сравнения; 6 - система обратной связи; 7, 7' - моторчики электродвигателей; 8 - барабан с длинами волн; 9 - светокомпенсатор
Верхний световой поток называется потоком сравнения, он проходит через кювету с растворителем. Нижний – основным (регистрирующим или индикаторным), он проходит через кювету с анализируемым раствором. В индикаторный световой поток введен светокомпенсатор (серые фильтры) - оптическая заслонка, которая ослабляет поток сравнения в такой же степени в какой растворенное вещество ослабляет индикаторный световой поток.
Лабораторная работа № 12 Выполнение работы 1) Спектрофотометр СФ-10 включает лаборант за 10-15 мин до начала работы. 2) Приготавливают стандартные растворы чистых красителей. Выбор красителей и их число определяет преподаватель (обычно анализируемая смесь состоит из трех красителей). Для каждого красителя приготавливают 4 стандартных раствора в мерных колбах вместимостью 50 см3. В предварительно вымытые и ополоснутые дистиллированной водой колбы добавляют по бюретке заданное лаборантом количество миллилитров исходного стандартного раствора красителя. Объем колб доводят дистиллированной водой до метки, убирают капли выше метки фильтровальной бумагой, закрывают пробкой, тщательно перемешивают. 3) Получают у лаборанта анализируемую смесь красителей в колбу на 50 см3. Доводят объем колбы дистиллированной водой до метки, закрывают пробкой, перемешивают. 4) Подготавливают две кюветы с одинаковой толщиной, моют водопроводной, ополаскивают дистиллированной водой. В одну кювету наливают дистиллированную воду, в другую - раствор красителя. 5) Устанавливают кюветы в специальных держателях в кюветное отделение спектрофотометра. Кювету с дистиллированной водой справа, с раствором – слева. 6) Закрепляют специальный бланк на барабане спектрофотометра и записывают спектры поглощения всех стандартных и анализируемого растворов на одном бланке. Порядок записи спектров поглощения смотри в разделе 3.6. 7) По окончании записи спектров, кюветы моют, ополаскивают дистиллированной водой и оставляют в спектрофотометре. Приводят рабочее место в порядок.
Расчет результатов анализа
1) Расчет концентрации приготовленных стандартных растворов красителей
где Сисх,i., Ск,i – концентрация исходного стандартного раствора красителя и в колбе соответственно, моль/дм3; Vисх. – объем исходного стандартного раствора, отмеренный по бюретке, дм3; Vк – объем колбы, дм3; 2) Выбор аналитических длин волн. Число аналитических длин волн соответствует числу красителей в анализируемой смеси (для трехкомпонентной смеси: lа1, lа2, lа3). Обычно аналитическая длина волны выбирается в максимумах полос поглощения чистых красителей, в каждом конкретном случае для смеси красителей необходимо учитывать пересечение спектров поглощения чистых компонентов (выбор аналитических длин волн следует согласовать с преподавателем). 1) Определение коэффициентов экстинкции на аналитических длинах волн. Таблица 1 - Результаты экспериментов
По результатам, приведенным в таблице 1, строят калибровочные графики в осях «абсорбция – концентрация». Если в результате построения получаются прямые линии, значит, закон Ламберта-Бугера-Бера выполняется. Коэффициенты экстинкции находят как тангенсы угла наклона прямых, равные отношению соответствующих катетов: e =А/С × l. При этом все величины следует брать в соответствующих единицах. Из графиков можно найти все девять коэффициентов экстинкции, некоторые из которых могут оказаться равными нулю, результаты расчетов заносят в таблицу 2.
Таблица 2 - Значения коэффициентов экстинкции
5) Расчет концентрации красителей в анализируемой смеси. Анализ смеси невзаимодействующих красителей, полосы поглощения которых пересекаются основан на аддитивностти абсорбций (1): Для выбранных аналитических длин волн записывают три уравнения (2):
Предполагая, что для каждого компонента смеси в рабочем интервале концентраций на всех выбранных длинах волн l а1, l а2, l а3 выполняется закон Ламберта-Бугера-Бера, получаем систему уравнений (3), в которую входят искомые концентрации красителей СI, СII, СIII.
Чтобы воспользоваться системой уравнений (3), необходимо измерить на бланке абсорбцию анализируемой смеси красителей на выбранных длинах волн, подставить значения коэффициентов ослабления из таблицы 2 и учесть толщину кюветы. Если на некоторой длине волны поглощает только один из компонентов смеси, то его концентрацию можно найти по калибровочному графику. Учитывая эти особенности, можно упростить расчет концентрации, выбрав соответствующим образом аналитические длины волн. Достоверность полученных результатов проверяют у преподавателя или лаборанта, оформляют отчет по лабораторной работе.
Вопросы к отчету по работе 1 Метод абсорбционной спектроскопии, его сущность и область применения. 2 Электромагнитный спектр, соотношение между длиной волны (l), частотой (n) и волновым числом ( 3 Спектры поглощения, способы их изображения. Применение спектров поглощения в аналитической химии. 4 Схема простейшего монохроматора, диспергирующая призма и дифракционная решетка. 5 Запись спектров поглощения на однолучевом и двухлучевом спектрофотометре. 6 Качественный и количественный анализ по электронным спектрам поглощения. Закон Ламберта-Бугера-Бера 7 Особенности количественного анализа смесей красителей с пересекающимися и непересекающимися спектрами. Правило аддитивности абсорбций.
ПЛАМЕННАЯ ФОТОМЕТРИЯ Теоретические основы
Метод пламенной фотометрии является одним их видов эмиссионной спектроскопии. Он основан на измерении интенсивности излучения, испускаемом анализируемыми элементами в пламени. Чувствительность метода определяется температурой пламени и условиями возбуждения атомов. В качестве источников возбуждения атомов используется пламя различных типов. Средняя температура некоторых наиболее широко распространенных видов источников возбуждения приведена в таблице 5.1.
Таблица 5.1 - Основные источники возбуждения атомов
Вследствие невысокой температуры пламени горючих смесей, их эмиссионные спектры состоят их небольшого числа спектральных линий и имеют низкие потенциала возбуждения. Это позволяет использовать при анализе фотометры, снабженные интерференционными светофильтрами. Атомные спектральные линии в пламени излучают щелочные и щелочноземельные металлы, галлий, кадмий, хром, марганец, никель, кобальт, медь, серебро и другие элементы. В настоящее время методом пламенной фотометрии можно определять до 40 элементов. Набор частот, соответствующий спектру каждого элемента, определяется структурой внешней оболочки атома и, следовательно, положением элемента в таблице Менделеева. Вследствие этого не может быть случаев полного совпадения эмиссионных спектров различных веществ. Качественный анализ анализируемых веществ основан на наличии в спектре излучения характеристических линий, присущих данному элементу. В пламенной фотометрии качественный анализ проводится с использованием набора светофильтров. В основе количественного анализа лежит прямо пропорциональная зависимость интенсивности спектральных линий от числа возбужденных частиц, и, следовательно, от концентрации определяемого элемента при постоянных температуре и условиях атомизации. Эта зависимость выражается уравнением Ломакина: где I – интенсивность спектральной линии; с – концентрация элемента в пробе; a – коэффициент, зависящий от режима работы источника возбуждения; b – коэффициент самопоглощения. Определяемые элементы поступают в пламя в виде аэрозоля, полученного при распылении анализируемых растворов воздухом в распылительной камере. В пламени протекает ряд следующих процессов: 1) Растворитель испаряется или сгорает, а в пламени остаются мельчайшие частицы твердого образца; 2) Твердые вещества переходят в газообразное состояние (превращаются в пар), то есть образуется атомный пар; 3) Возбуждение атомов под действием энергии пламени. Возбуждение газообразных атомов связано с переходом одного из электронов в более высокое энергетическое состояние; В условиях термодинамического равновесия доля возбужденных атомов определяется уравнением Больцмана: где N* – количество возбужденных атомов в пламени; N0 – общее количество атомов в пламени; ΔE – потенциал возбуждения, Дж; k – постоянная Больцмана (k=1,380662 10-22 Дж К); T – абсолютная температура, К. Согласно уравнению Больцмана, только небольшая часть газообразных атомов переходит в возбужденное состояние и доля возбужденных атомов быстро снижается при возрастании потенциала возбуждения элементов. Например, при одинаковой температуре доля возбужденных атомов натрия в 107 раз превышает долю возбужденных атомов цинка. Вследствие этого для определения цинка требуется более высокая температура. При повышении температуры число возбужденных атомов увеличивается. 4) Излучение света возбужденными атомами: при возвращении электрона из возбужденного состояния на более низкий энергетический уровень или в основное состояние испускается ультрафиолетовое или видимое излучение. Интенсивность и частота излучения зависят от природы, концентрации анализируемого элемента в растворе. Частота испускаемого излучения зависит от разности энергий двух состояний атома: где E2 – энергия более высокого возбужденного состояния, Дж; Е1 – энергия более низкого возбужденного или основного состояния, Дж; h – постоянная Планка; ν – частота испускаемого ультрафиолетового или видимого света, Гц; с – скорость света, м/с; λ – длина волны излучаемого света, м.
Схема пламенного фотометра Схема пламенного фотометра представлена на рисунке 5.1.

Рисунок 5.1 - Схема пламенного фотометра ПФЛ-1: 1 – стаканчик с раствором; 2- распылитель; 3 – распылительная камера; 4 – водяной затвор; 5 – смесительная камера; 6 – горелка; 7 – пламя; 8 – входная щель; 9 – светофильтр; 10 – фотоэлемент; 11 – усилитель; 12 – гальванометр В качестве монохроматора в приборе использованы три сменных светофильтра, полосы пропускания которых соответствуют аналитическим спектральным линиям натрия, калия и кальция [1, табл. 8]. Полосы пропускания светофильтров являются достаточно узкими. Светофильтры пропускают излучение аналитической линии определяемого элемента и поглощают излучение других элементов. В качестве аналитического сигнала при количественном анализе в пламенной фотометрии используются показания гальванометра 12, расположенного на передней панели прибора. Гальванометр измеряет ток фотоэлемента 10, который зависит от интенсивности спектральной линии и, в свою очередь, от концентрации анализируемого раствора.
Лабораторная работа № 15 ОПРЕДЕЛЕНИЕ ЭЛЕМЕНТОВ Выполнение работы:
1) Готовят серии стандартных растворов солей каждого определяемого элемента с различными фиксированными концентрациями, внося в мерные колбы на 50 см3 заданный лаборантом объем исходного раствора. Объем растворов доводят дистиллированной водой до метки. Растворы в колбах тщательно перемешивают. 2) Пробу для анализа студенты получают у лаборанта в мерной колбе и также доводят до метки дистиллированной водой. 3) Заполняют нумерованные стаканчики приготовленными стандартными, анализируемыми растворами и дистиллированной водой. 4) Устанавливают светофильтр, соответствующий анализируемому элементу, поворачивая рычаг на правой стенке прибора. 5) Трубку распылителя опускают в стаканчик с дистиллированной водой и с помощью ручки гальванометра, расположенной на передней панели прибора слева, устанавливают стрелку гальванометра на ноль. 6) Трубку распылителя опускают в стаканчик с самым концентрированным раствором, при этом стрелка прибора отклоняется вправо. С помощью правой ручки гальванометра устанавливают стрелку в положение, соответствующее делению шкалы 80-90. Повторяют процедуру 3-4 раза. Такая методика гарантирует от зашкаливания прибора при дальнейших измерениях, т.к. все растворы с меньшей концентрацией соответствуют показаниям прибора между нулем и максимальным показанием гальванометра. 7) Снимают показания гальванометра для остальных растворов анализируемого элемента в порядке снижения концентрации. Затем снимают показания гальванометра анализируемой смеси. Результаты заносят в таблицу 1. 8) По полученным экспериментальным данным строят калибровочный график. Для этого по оси абсцисс откладывают значения концентрации растворов в единицах мг/мл, по оси ординат – соответствующие значения показаний гальванометра. 9) По калибровочному графику определяют концентрацию ионов анализируемого металла. 10) Повторяют измерения с п.3 по п.7 для других солей. Для непрерывного контроля результатов рекомендуется наносить значения концентрации и показания прибора на калибровочный график одновременно с записью в таблицу. В этом случае можно проверить точки, выпавшие из графической зависимости, вновь установив стаканчик с раствором для измерения.
Расчет результатов анализа 1) Расчет концентрации приготовленных стандартных растворов солей
где Сисх.ст. р-ра – концентрация исходного стандартного раствора соли в склянке, мг/мл; Сст. – концентрация, приготовленного раствора в колбе, мг/мл; Vисх.ст.р-ра – объем исходного стандартного раствора, отмеренный по бюретке, мл; Vк – объем колбы, мл. 2) Экспериментальные результаты заносят в таблицу 1.
Таблица 1 – Результаты эксперимента
Построение калибровочного графика и определение концентрации металла в анализируемом растворе. Калибровочный график строится по результатам, приведенным в таблице 1.

Результат проверяют у преподавателя или лаборанта, оформляют отчет по выполненной работе. Вопросы к отчету по лабораторной работе
1 Принципы анализа, основанного на спектрах испускания. Природа линейчатых спектров. Атомный пар. 2 Различные способы возбуждения атомов. 3 Метод пламенной фотометрии, область его применения. 4 Назначение светофильтров, качественный анализ 5 Методы количественного анализа (метод калибровочного графика, метод внутреннего стандарта, метод добавок). 6 Схема прибора и принцип его действия. Дополнительные вопросы 1 Устройство газовых горелок. 2 Структура пламени, процессы, протекающие в пламени. 3 Основные характеристики атомных спектральных линий: частота, интенсивность, ширина.
БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1 Основы аналитической химии: в 2 т. / под ред. Ю.А. Золотова. – М. : Высша. шк., 1996. 2 Крешков, А.П. Основы аналитической химии. Т. 2, 3. – 2-е изд. – М. : Химия, 1965. 3 Васильев, В.П. Аналитическая химия. Ч. 1, 2.– М. : Высшая школа, 1989. 4 Аналитическая химия / по ред.проф. О.М. Петрухина. - М. : Химия, 1993. 5 Алексеев, В.Н. Количественный анализ. – 4-е изд. – М. : Химия, 1972. 6 Скуг., Уэст Д. Основы аналитической химии. Т.2. – М. : Мир, 1979. 7 Ушакова, Н.Н. Курс аналитической химии. – М. : Изд-во Моск. ун-та, 1987. 8 Лурье, Ю.Ю. Справочник по аналитической химии. – 5-е изд. – М.: Химия, 1979. 9 Метрологические характеристики анализа вещества. Аттестация аналитических методик: Метод. указания к лабораторным работам / Cост. Ф.П. Черняковский; Яросл. политехн. ин-т. – Ярославль, 1988. – 24 с. – МУ № 1221. 10 Лабораторный практикум по физико-химическим методам анализа: Учеб. пособие / Под ред. проф. Ф.П. Черняковского. – 3-е изд., испр. и доп.; Яросл. политехн. ин-т. – Ярославль, 1990. – 82 с. – МУ № 1455. 11 Черняковский Ф.П. Основы физико-химических методов анализа и исследования органических веществ: Учеб. пособие / Яросл. политехн. ин-т. – 5-е изд., испр. и доп. – Ярославль, 1987. – 90 с. – МУ № 1177. 12 Методические указания и контрольные задания по аналитической химии для студентов 4-го курса заочного обучения. Физико-химические методы анализа / Сост.: Ф.П. Черняковский, О.В. Кузьмичев, Л.А. Шмулевич, С.И. Зайцева, Н.С. Кичева; Яросл. гос. техн. ун-т. – Ярославль, 1997. – 64 с. – МУ № 2138. 13 Методические указания для подготовки к коллоквиумам по аналитической химии для студентов дневного отделения химико-технологического факультета / Сост. : О.В. Кузьмичев, Е.А. Сапунов, Л.А. Шмулевич, Н.С. Кичева, В.Н. Крутецкая, С.И. Зайцева; Яросл. гос. техн. ун-т. – Ярославль, 2002. – 32 с. – МУ №2346. 14 Кросс, А.Д. Введение в практическую инфракрасную спектроскопию. – М. : Изд-во иностр. лит-ра,1961. 15 Клаузен, Н.А. Атлас инфракрасных спектров каучуков и некоторых инградиентов резиновых смесей / Н.А. Клаузен, Л.Н. Семенова. – М.-Л. : Изд-во Химия, 1965.
Теоретические основы
Хроматография – это совокупность методов разделения и анализа сложных смесей веществ, основанных на распределении веществ между двумя фазами – подвижной (газ или жидкость) и неподвижной (жидкость или твёрдое вещество). Хроматография – гибридный аналитический метод, сочетающий разделение и определение веществ в одном приборе – хроматографе, содержащем хроматографическую колонку и детектор. Метод позволяет разделять многокомпонентную смесь, идентифицировать компоненты и определять её количественный состав. В основу классификации многочисленных хроматографических методов положены следующие признаки: агрегатное состояние подвижной и неподвижной фаз (ГЖХ, ГТХ, ЖТХ, ЖЖХ); механизм взаимодействия сорбент-сорбат (распределительная, ионообменная, адсорбционная и др.); форма слоя сорбента (колоночная, бумажная, тонкослойная); цель хроматографирования (аналитическая, препаративная, промышленная); по способу получения хроматограмм (элюентная, вытеснительная, фронтальная). В настоящее время имеются десятки разновидностей хроматографических методов анализа, которые продолжают совершенствоваться и развиваться. Анализируемой смеси
Качественной характеристикой каждого компонента в газо-жидкостной хроматографии является время удерживания (t) или удерживаемый объём (объём газа-носителя, необходимый для вымывания компонента пробы), которые не зависят ни от количества введённой пробы, ни от чувствительности детектора. Качественный анализ основан на сравнении времён удерживания известных чистых веществ с временами удерживания компонентов пробы. Типичная хроматограмма трёхкомпонентной смеси представлена на рисунке 1.1. На хроматограмме времена удерживания пропорциональны отрезкам от точки начального отброса самописца (момент ввода пробы) “а” до t1, t2, t3.
Количественный анализ
Количественный анализ в газо-жидкостной хроматографии может быть проведён различными методами (приёмами): Метод нормализации площадей Метод нормализации площадей может быть использован только в том случае, если все компоненты смеси дают хорошо записанные хроматографические пики и имеют различные времена удерживания. Аналитическим сигналом в газо-жидкостной хроматографии является площадь под хроматографическим пиком. Однако площадь
Рисунок 1.1 – Типичная хроматограмма трехкомпонентной смеси: а – начальный отброс пера самописца; t1, t2, t3 – отрезки, пропорциональные временам удерживания компонентов смеси
под пиком компонента пробы зависит не только от его содержания в пробе, но и от объёма введённой пробы, а также от чувствительности детектора к компонентам пробы. Для этого необходимо ввести в расчёт соответствующие поправки. Влияние объёма введённой пробы можно исключить, применяя приём расчёта, основанный на измерении относительных площадей под пиками. Опытным путём установлено, что даже при широком варьировании объёма пробы отношения площадей под пиками для любой пары компонентов остаются неизменными. Поэтому при расчёте определяют относительные площади как отношение площади данного пика к сумме площадей всех пиков пробы Определение площади хроматографического пика Площадь хроматографического пика S определяют как площадь треугольника по формуле: S = h d , где h – высота пика (рисунок 1.2) от базовой линии до вершины, мм; d – отрезок прямой, параллельной базовой (нулевой) линии на высоте
Рисунок 1.2 – Параметры хроматографического пика: h – высота пика от базовой линии до вершины; d – ширина пика Метод стандартных добавок Метод стандартной добавки основан на том, что в навеску контрольной смеси вносят точную навеску анализируемого вещества, присутствующего в контрольной смеси, и снимают хроматограммы исходной контрольной смеси и контрольной смеси с внесённой в неё стандартной добавкой. Методика анализа. В предварительно взвешенную колбочку с пришлифованной пробкой вносят пипеткой около 2 см3 контрольной смеси ( Далее снимают хроматограммы исходной контрольной смеси и контрольной смеси с внесённой в неё стандартной добавкой определяемого компонента. Измеряют на хроматограммах площади под пиком анализируемого компонента и рассчитывают результат анализа по формуле
где S х – площадь под пиком анализируемого компонента в пробе; S х+ст – площадь под пиком анализируемого компонента в пробе после введения в пробу его стандартной добавки Сст; С(х) – концентрация анализируемого компонента в пробе; Сст – концентрация стандартной добавки анализируемого компонента, %:
где m доб – масса добавки, г; m пробы – масса хроматографируемой пробы, г.
Рисунок 1.3 – Калибровочный график
Концентрацию i-го компонента в пробе (%) рассчитывают по формуле
где m пробы – масса хроматографируемой пробы, г; m i – содержание i-го компонента, найденное по калибровочному графику (см. рисунок 1.3), г. Лабораторная работа № 1 Расчет результатов анализа 1) Результаты измерений площадей пиков (высот пиков, полуширины пиков), расчета калибровочных коэффициентов (формула (1.3)) и концентраций компонентов в анализируемой пробе методами нормализации площадей (формула (1.4)), внутреннего стандарта (формула (1.5)) или стандартной добавки (формула (1.6)) заносят в таблицу 1 и проверяют результат у преподавателя (лаборанта).
Таблица 1 – Результаты измерений
2) Далее сопоставляют метрологические показатели каждого из рассмотренных методов анализа. При этом, в случае аккуратного выполнения всех рекомендованных процедур, при использовании одного и того же прибора, в тождественных условиях и однотипных приёмов измерения сигнала детектора, должны прийти к выводу, что методы нормализации площадей, внутреннего стандарта и стандартной добавки по воспроизводимости и правильности результатов равноценны. Сравнивают также и другие параметры методов: трудоёмкость, длительность и др. и формулируют краткие выводы по проделанной работе.
Вопросы к отчету по лабораторной работе: «Качественный и количественный анализ многокомпонентных смесей углеводородов методом газо-жидкостной хроматографии»
1 Теоретические основы и области применения хроматографии. Классификация хроматографических методов. 2 Газо-жидкостная хроматография, теоретические основы и область её применения. 3 Блок-схема газо-жидкостного хроматографа. 4 Качественный и количественный анализ в газо-жидкостной хроматографии. 5 Приёмы количественного хроматографического анализа: метод нормализации площадей, метод внутреннего стандарта, метод добавок, метод абсолютной калибровки. 6 Детектирование в газо-жидкостной хроматографии. Устройство и принципы действия детектора катарометра и пламенно-ионизационного детектора. 7 Механизм разделения компонентов в хроматографии. Закон распределения Нернста.
Теоретические основы
Электропроводность раствора электролита G и его концентрация С связаны между собой функциональной зависимостью. При низких концентрациях где k – некоторая постоянная величина. Для правильного построения методики кондуктометрического анализа необходимо знать электрические свойства растворов, термины, определения, физико-химические основы переноса зарядов в электролитах, причины нелинейности электропроводности [3]. Электропроводность растворов в системе СИ G измеряется в сименсах (См). Различают удельную χ
где С – молярная концентрация эквивалента, моль/дм3 . Эквивалентная электропроводность – аддитивная величина, которая выражается соотношением
где α – степень диссоциации; z – заряд иона;
В свою очередь, Таблица 2.1 - Подвижности ионов в водных растворах при бесконечном разбавлении при t =25˚С
Подвижности ионов определяют значение k в (2.1) и вид кривых титрования. Измерение электропроводности растворов и величин, связанных с электропроводностью, лежит в основе кондуктометрического метода анализа. Метод реализуется двумя вариантами применения. Прямая кондуктометрия, основанная на измерении электропроводности растворов по уравнению (2.1). Аналитическим сигналом является G, а k – коэффициент чувствительности. В данном методе необходимо знать функциональную зависимость G от C. Ее устанавливают, измеряя электропроводность стандартных растворов с известными концентрациями, и выражают в виде калибровочной характеристики: в виде графика, таблицы или информации, введенной в ЭВМ. Кондуктометрическое титрование, в котором по изменению электропроводности анализируемого раствора определяют точку эквивалентности. В кондуктометрическом титровании вид функциональной зависимости не имеет существенного значения, важен перелом на кривой кондуктометрического титрования, соответствующий точке эквивалентности.
Рисунок 2.1 – Калибровочная кривая метода прямой кондуктометрии
Рисунок 2.3 - Кривая кондуктометрического титрования смеси кислот
В этом случае первым титруется сильный протолит, электропроводность значительно понижается, затем титруется слабый электролит, электропроводность незначительно повышается за счет накопления ионов с невысокой подвижностью (катионов и анионов). После достижения второй точки эквивалентности электропроводность раствора увеличивается вследствие возрастания концентрации ионов OH- или (H3O+) с высокой подвижностью.
Рисунок 2.4 – Схема моста Уитстона
Рисунок 2.5 – Схема сосуда с прочно закрепленными платиновыми электродами (кондуктометрическая ячейка) Таблица 2.2 – Экспериментальные данные
Затем строят кривую титрования, откладывая на оси ординат электропроводность (см. рисунок 2.2). На оси абсцисс – объем титранта. Точка эквивалентности определяется графически на пересечении прямых. Зная объем и концентрацию титранта, рассчитывают содержание анализируемого вещества.
Высокочастотное титрование Теоретические основы
Высокочастотное титрование является вариантом бесконтактного кондуктометрического метода анализа, в котором исследуемый раствор подвергают действию электрического поля высокой частоты (порядка нескольких МГц). Под действием поля переменного тока обычных частот ионы в растворе колеблются около некоторого состояния равновесия. По мере увеличения частоты внешнего электрического поля электропроводность растворов электролитов увеличивается, поскольку уменьшается амплитуда колебаний ионов в поле переменного тока. Поле высокой частоты деформирует молекулу, поляризуя ее (деформационная поляризация) и заставляет полярную молекулу определенным образом перемещаться (ориентационная поляризация). Оба типа поляризации приводят к возникновению кратковременных токов, изменяющих электропроводность, диэлектрические свойства и магнитную проницаемость растворов Полная электропроводность высокочастотной кондуктометрической ячейки λ складывается из активной составляющей λакт. – истинной проводимости раствора – и реактивной составляющей λреакт. – мнимой электропроводности, зависящей от частоты и тока ячейки:
Функциональная зависимость этих параметров от состава растворов сложна и не может быть использована для прямого высокочастотного анализа. Широкое распространение получил метод высокочастотного титрования с использованием реакций кислотно-основного взаимодействия, осаждения, окисления-восстановления, компексообразования. При высокочастотном титровании измеряют электрические (и магнитные) параметры исследуемых растворов в зависимости от их состава. Форма кривой высокочастотного титрования зависит от частоты налагаемого электрического поля, состава раствора, концентрации титранта, типа ячейки. Точка эквивалентности на кривой титрования должна располагаться на изломе кривой, который находится на пересечении прямолинейных участков. Высокочастотное титрование проводится в электролитических ячейках, в которых исследуемый электролит не контактирует с электродами, что исключает поляризацию электродов и химическое взаимодействие материала электродов с раствором. Материал электродов не влияет на результаты анализа. К достоинствам метода относится также возможность анализа агрессивных сред, паст, эмульсий, неводных растворов. Метод высокочастотного титрования позволяет проводить определения с погрешностью до 2 %. Метод не избирателен, т.к. проведению анализа мешают все посторонние ионы в растворе.
Теоретические основы
Потенциометрические методы анализа основаны на использовании зависимости ЭДС электрохимической ячейки от активности определяемого иона в растворе. Простейшая электрохимическая ячейка содержит два электрода: индикаторный, потенциал которого прямо или косвенно зависит от концентрации потенциалопределяемого иона, и электрода сравнения. ЭДС = Е1 – Е2, (2.2), где ЭДС – электродвижущая сила, Е1 и Е2 – потенциалы электродов, индикаторного и сравнения соответственно. Математическое выражение зависимости электродного потенциала от активной концентрации ионов, относительно которых обратим электрод, даётся уравнением Нернста:
где Е0 – стандартный потенциал системы; R – газовая постоянная (8,312 Дж/моль·К); T – абсолютная температура; F – постоянная Фарадея (96500 Кл/моль); п – число электронов, участвующих в электродной реакции; a – активная концентрация определяемого иона.
Рисунок 2.8 - Принципиальная схема измерения потенциала: 1 – источник с известной ЭДС (аккумулятор); 2 – постоянное сопротивление; 3 – реохорд 4 – электролитическая ячейка; 5 – амперметр; 6 – вольтметр
Потенциометрический метод анализа подразделяется: 1) на прямую потенциометрию (ионометрия); 2) потенциометрическое титрование в водных и неводных средах.
Рисунок 2.10 - Измерительная схема с использованием ионоселективных электродов: 1 – ионоселективный электрод; 2 – электрод сравнения; 3 – анализируемый раствор; 4 – измерительный прибор
В зависимости от применяемого электродноактивного материала ионоселективные электроды разделяются на электроды с твёрдыми и жидкими мембранами. Электродами с твёрдыми мембранами являются стеклянные электроды, электроды с кристаллической мембраной и гомогенные и гетерогенные осадочные мембранные электроды. Конструктивно все ионоселективные электроды имеют много общего (рисунок 2.11).
Рисунок 2.11 - Конструкции различных ионоселективных электродов: а - стеклянный электрод, б - электрод с твёрдой мембраной, в - электрод с жидкой мембраной; 1 – мембрана; 2 – внутренний электрод сравнения (AgCl/Аg); 3 – внутренний (стандартный) раствор; 4 – жидкий ионообменник Твёрдая мембрана представляет собой малорастворимое соединение, обладающее ионной проводимостью. Электроды с такой мембраной “откликаются” на ионы, переносимые через мембрану, и на противоионы, образующие с первыми малорастворимое соединение мембраны, так как концентрации этих ионов в растворе связаны между собой произведением растворимости. Твёрдой мембраной может быть монокристалл с ионной проводимостью при комнатной температуре. Перенос заряда в кристалле происходит за счёт дефектов кристаллической решётки, когда вакансии заполняются только определёнными видами ионов, идеально соответствующими определённому иону в отношении размера, формы и распределения заряда. Посторонние ионы не могут войти в кристалл и перемещаться в нём. Если мембрана не обладает механической прочностью, её вводят в инертную матрицу (каучук, полистирол и т.д.) – такая мембрана называется гетерогенной. Электроды с твёрдыми мембранами: фторид-селективный на основе электродноактивного соединения LaF3, сульфид-селективный на основе Ag2S, Cl-селективный на основе Ag2S + AgCl и т. д. Жидкие мембраны представляют собой не смешивающуюся с водой органическую жидкость (растворитель с растворённым в нём ионообменным веществом), которая обладает селективным свойством проникновения через неё различных ионов. Жидкая мембрана находится в контакте с двумя водными растворами и должна быть практически нерастворима в них. Этим требованиям отвечают органические вещества с низкой диэлектрической проницаемостью и большим молекулярным весом. В качестве жидких мембран применяют органические ионообменники (четвертичные аммониевые и фосфониевые основания), нейтральные носители и биологически активные вещества (валиномицин, нонактин и др.). Все эти электроды высокоизбирательны и широко используются в практике. Важной характеристикой ионоселективного электрода является его коэффициент селективности (избирательности), показывающий во сколько раз электрод более чувствителен к данным ионам, чем к посторонним (мешающим). Мешающее влияние ионов можно оценить с помощью коэффициента селективности по уравнению Никольского: где i – потенциалопределяющий ион, j – мешающий ион,
Электрод должен иметь низкие значения селективности. В настоящее время имеются ионселективные электроды для определения ионов F-, Cl-, Br-, I-, CN-, S-, Ag+, SCN-, Cu2+, Pb2+, Cd2+, NO3-, K+, Ca2+, Mg2+ и др. Метод ионометрии обладает рядом преимуществ перед другими методами анализа. Метод прост, удобен, требует малое количество времени для определения анализируемого вещества, позволяет проводить измерения ЭДС чрезвычайно малых проб, меньше 1 см3 и без разложения анализируемого раствора. Но селективность созданных электродов ещё недостаточно высока, а возможность создания электродов, чувствительных к многозарядным ионам, ограничена точностью измерения ЭДС, так как при 10-кратном изменении активности для однозарядных ионов изменение потенциала составляет 59,16 мВ, для двухзарядных – 28,56 мВ, для трёхзарядных – 19,72 мВ. Мембранные ионоселективные электроды имеют большое сопротивление, и для измерения ЭДС с мембранными электродами применяют потенциометры на основе усилительных схем. Для этой цели служат ионометры и рН-метры различных марок (рН-340, рН-121, рН-262, рН-673 и др.). Так как применение расчётных методов определения концентраций ионов (по уравнению (2.3)) сопряжено со значительными трудностями, то в ионометрии пользуются двумя методами относительных измерений с использованием стандартных растворов: 1 – метод градуировочного графика, 2 – метод стандартных добавок.
Рисунок 2.12 – Нахождение точки максимального перегиба Лабораторная работа № 4 И БОРНОЙ КИСЛОТ МЕТОДОМ Расчет результатов анализа
По результатам кондуктометрического титрования раствора хлористоводородной кислоты рассчитывается молярная концентрация эквивалента щелочи по формуле
Расчет содержания кислот в смеси производят по формуле
где C (NaOH) и V (NaOH) – молярная концентрация эквивалента титранта и объем титранта, затраченного на титрование кислоты;
Лабораторная работа № 9 Выполнение работы
1. Потенциометр включают в электрическую сеть. Тумблер включения ставят в положение “ВКЛ.”. прибор прогревается в течение 15 мин. Выключают прибор только после окончания работы. Переключатель “Виды работ” ставят в положение “мВ”. 2. Сухой стакан для титрования взвешивают на аналитических весах, затем повторяют взвешивание с навеской бензойной кислоты (0,05-0,07 г), по результатам взвешивания рассчитывают точную навеску. 3. Навеску растворяют в 20 см3 ацетона. В стакан для титрования аккуратно опускают мешалку и погружают электроды в раствор так, чтобы шарик стеклянного электрода находился на расстоянии не менее 5 см3 от мешалки. Если при этом покрыто менее половины стеклянного шарика, то для погружения шарика следует добавить в стакан воды. 4. Промыть бюретку дистиллированной водой, затем раствором титранта. После этого заполнить бюретку титрантом до нулевого деления при заполненном кончике бюретки. 5. В начале процесса титрования бензойной кислоты титрант (КОН) добавляют (до 2 см3) порциями по 0,5 см3, а затем – по 0,2 см3. После скачка потенциала в точке эквивалентности необходимо сделать ещё 6-7 замеров рН, добавляя титрант по 0,2 см3. 6. Проводят титрование смеси кислот, указанной преподавателем. В стакан для титрования получают анализируемую смесь, добавляют 20 см3 дистиллированной воды и проводят титрование: сначала ориентировочное, добавляя титрант по 0,5 см3, затем точное, добавляя титрант по 0,2 см3. 7. Помещают анализируемую смесь в стакан для титрования и проводят неводное титрование. Для этого в смесь вводят 20 см3 ацетона (проверить правильность расположения электродов) и титруют аналогично п.6.
Расчет результатов анализа
1) Определение концентрации раствора титранта (КОН).
Таблица 1 – Результаты эксперимента
На миллиметровке вычерчивают кривую титрования в координатах потенциал Е (мВ) – объём титранта V (см3). Находят на ней точку эквивалентности как точку перегиба, пользуясь методом трёх касательных, или более точно – как точку, соответствующую максимальной степени крутизны кривой титрования. По точному объёму КОН, израсходованному на титрование до точки эквивалентности, рассчитывают концентрацию титранта:
где mб.к. – навеска бензойной кислоты, г; Mб.к. – молярная масса эквивалента бензойной кислоты, Mб.к.=122,12; V – объём щёлочи, израсходованный на титрование, см3. 2) Определение суммарного содержания кислот в анализируемой смеси.
Таблица 2 – Результаты определения суммарного содержания кислот Вопросы к отчету по теме «Потенциометрический метод анализа» 1 На чём основан метод потенциометрии? 2 Какая зависимость выражается уравнением Нернста? 3 Каким требованиям должны отвечать электроды, применяемые в потенциометрии? 4 В чём сущность потенциометрического определения рН раствора? Какие индикаторные электроды могут использоваться для определения рН? 5 Как устроен стеклянный электрод? Каковы его достоинства и недостатки? Как можно определить стандартный потенциал этого электрода? 6 Каковы достоинства, недостатки и области применения прямой потенциометрии? 7 Каковы особенности потенциометрического титрования в неводных средах? Какие требования предъявляются к неводному растворителю? 8 Достоинства и области применения потенциометрического титрования в неводных средах.
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Отчёт о лабораторной работе по дисциплине «Физико-химические методы анализа»
ЯГТУ 18.03.01 – 001 ЛР СОГЛАСОВАНО
Руководитель . хим. наук, доцент канд. экон. наук, доцент ____________С. А. Соловьева ____________М. А. Угрюмова
Отчёт выполнил студент группы ЗО-30 __________Ю.Б. Соловьёв 27.02.2017
2017
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Последнее изменение этой страницы: 2019-04-20; Просмотров: 1750; Нарушение авторского права страницы
 , (1.1)
, (1.1) ,
, 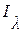 – интенсивность падающего и прошедшего через раствор света соответственно, Лм; 10 – основание десятичного логарифма; e – молярный коэффициент ослабления, дм3×моль-1×см-1; С – концентрация раствора, моль/дм3; l – толщина слоя поглощающего раствора, см.
– интенсивность падающего и прошедшего через раствор света соответственно, Лм; 10 – основание десятичного логарифма; e – молярный коэффициент ослабления, дм3×моль-1×см-1; С – концентрация раствора, моль/дм3; l – толщина слоя поглощающего раствора, см. , (1.2)
, (1.2) является важной оптической характеристикой веществ, ее называют абсорбцией и обозначают А:
является важной оптической характеристикой веществ, ее называют абсорбцией и обозначают А: . (1.3)
. (1.3) , (1.4)
, (1.4) – абсорбция смеси невзаимодействующих красителей на аналитической длине волны
– абсорбция смеси невзаимодействующих красителей на аналитической длине волны  ;
; – абсорбции отдельных красителей, измеренные на той же длине волны и в тех же кюветах, что и абсорбция смеси.
– абсорбции отдельных красителей, измеренные на той же длине волны и в тех же кюветах, что и абсорбция смеси.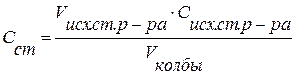 ,
,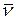 ). Спектры поглощения записывают на одно- или двухлучевых спектрофотометрах.
). Спектры поглощения записывают на одно- или двухлучевых спектрофотометрах.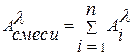 [2]. В случае, если компоненты смеси взаимодействуют между собой, то количественный анализ следует проводить с учетом закона аддитивности и условий, определяющих химическое взаимодействие между компонентами.
[2]. В случае, если компоненты смеси взаимодействуют между собой, то количественный анализ следует проводить с учетом закона аддитивности и условий, определяющих химическое взаимодействие между компонентами. ,
, (1)
(1)
 (2)
(2)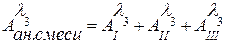

 (3)
(3)
 ).
). , (5.1)
, (5.1) , (5.2)
, (5.2) , (5.3)
, (5.3) ,
,
 .
.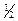 h, замеренный от внешней стороны одной боковой линии пика до внутренней стороны другой, мм. Измерения проводят с помощью специальной измерительной лупы.
h, замеренный от внешней стороны одной боковой линии пика до внутренней стороны другой, мм. Измерения проводят с помощью специальной измерительной лупы.
 800 мг) и взвешивают, а далее вносят одно из веществ (
800 мг) и взвешивают, а далее вносят одно из веществ (  100 мг), присутствующих в контрольной смеси (по указанию преподавателя), и вновь взвешивают.
100 мг), присутствующих в контрольной смеси (по указанию преподавателя), и вновь взвешивают.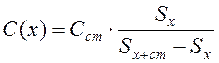 , (1.6)
, (1.6) , (1.7)
, (1.7) , (1.8)
, (1.8)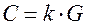 , (2.1)
, (2.1) и эквивалентную λ электропроводность раствора. Удельная электропроводность См/см или Ом-1·см-1 – электропроводность 1 см3 раствора, находящегося между электродами площадью 1 см2 каждый, расстояние между которыми равно 1 см. Эквивалентная электропроводность – электропроводность слоя электролита, содержащего 1 эквивалент вещества, измеренная при расстоянии между электродами 1 см. Удельная и эквивалентная электропроводность связаны соотношением
и эквивалентную λ электропроводность раствора. Удельная электропроводность См/см или Ом-1·см-1 – электропроводность 1 см3 раствора, находящегося между электродами площадью 1 см2 каждый, расстояние между которыми равно 1 см. Эквивалентная электропроводность – электропроводность слоя электролита, содержащего 1 эквивалент вещества, измеренная при расстоянии между электродами 1 см. Удельная и эквивалентная электропроводность связаны соотношением ,
,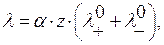
 и
и 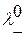 - подвижности положительных и отрицательных ионов.
- подвижности положительных и отрицательных ионов. ;
; 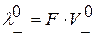 , где F – число Фарадея,
, где F – число Фарадея, 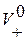 и
и  – абсолютные скорости движения ионов (в см/с) в поле напряженностью 1 В/см. В таблице 2.1 приведены подвижности ионов в водных растворах при бесконечном разбавлении при t=25˚С.
– абсолютные скорости движения ионов (в см/с) в поле напряженностью 1 В/см. В таблице 2.1 приведены подвижности ионов в водных растворах при бесконечном разбавлении при t=25˚С.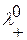
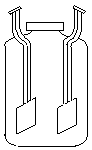
 .
.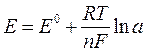 ,
,

 (2.4)
(2.4) может быть больше 1, тогда электрод “чувствует” мешающий ион больше, чем определяемый;
может быть больше 1, тогда электрод “чувствует” мешающий ион больше, чем определяемый; .
.
 - масса эквивалента определяемой кислоты.
- масса эквивалента определяемой кислоты. =36,5 (z=1);
=36,5 (z=1); 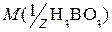 =61,83 (z=1).
=61,83 (z=1). ,
,