
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА. Кафедра «Химия»Стр 1 из 31Следующая ⇒
Кафедра «Химия»
Нечаева И.А. доцент кафедры «Биотехнология», к.б.н.
КОНСПЕКТ ЛЕКЦИЙ по дисциплине
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА ОБЪЕКТОВ ОКРУЖАЮЩЕЙ СРЕДЫ
Направление подготовки: 020100 Химия Профиль подготовки: Химия окружающей среды, химическая экспертиза и экологическая безопасность Квалификация (степень) выпускника: магистр Форма обучения: очная
Тула 2014 г.
Рассмотрено на заседании кафедры протокол №______ от «______» ________________20____г. Зав. кафедрой ___________________В.А. Алферов
Введение Глобальное загрязнение окружающей среды и неблагополучная экологическая ситуация в промышленных регионах обусловливают необходимость постоянного аналитического контроля (мониторинга) за загрязнением воздуха, качеством питьевой воды и накоплением токсичных химических веществ в почве. Острые проблемы экологического характера вызвали к жизни мощные общественные движения («Green Peace» и другие «зеленые» организации), подтолкнули правительства к заключению важных международных соглашений (Монреальский и Киотский протоколы). Произошли определенные изменения в массовом сознании. Приняты соответствующие изменения в законодательстве, открыты экологические специальности в высших учебных заведениях, появилось множество «экологических» книг и фильмов (иногда наивных и нелепых, иногда спекулятивных). Для объективного рассмотрения всех опасностей нужны и исключительно важны результаты химического анализа. Частыми и многочисленными стали конференции по методам и результатам анализа экологических объектов, например регулярный международный симпозиум по аналитической химии окружающей среды International Symposium on Environmental Analytical Chemistry. Нужны данные по содержанию углекислого газа, озона, канцерогенных органических соединений, оксидов серы и азота, радиоактивных изотопов и множества других микропримесей. Определять эти вещества нужно в широком концентрационном диапазоне. Из арсенала аналитической химии, насчитывающего более 150 методов, экологическая аналитическая химия использует наиболее эффективные и надежные методики, которые охватывают весь спектр загрязнений воздуха, воды, почвы, донных отложений и растительности – от газов и паров до твердых частиц и аэрозолей. Именно поэтому во многих ведущих ВУЗах России появились экологические специальности и специализации, стали готовить не только специалистов экологов-технологов, но и специалистов экологов-аналитиков. Кроме того, выпускники химических факультетов, получающих квалификацию «химик», должны иметь представление о принципах и методологии экологического контроля. Изложение материала базируется на базовых знаниях студентов по аналитической химии, которые развиваются и дополняются новыми сведениями. Экологическая аналитическая химия – это наука о выявлении и оценке источников и уровня загрязненности природных объектов вредными веществами в результате сбросов либо выбросов этих веществ в окружающую среду природопользователями, а также вследствие естественного образования и накопления. Почему вычленилась отдельная область аналитической химии – химия окружающей и природной среды? Можно назвать несколько причин оформления отдельного раздела аналитической химии: 1. Задача аналитической химии – поиск оптимальных решений для каждой аналитической задачи, взятой в отдельности. Например, определение содержания кадмия, свинца, цинка, хрома в сточной воде, бензпирена в присутствии ароматических и полиароматических углеводородов в воздухе. Реально лаборатория экоаналитического контроля должна контролировать одновременно десятки и сотни загрязнителей в разнородных объектах. Это требует от аналитика совершенно другого системного подхода к выбору конфигурации аналитической техники. Должна быть реализована методология получения массивов данных результатов химического анализа. В отсутствии такой методологии бессистемно приобретаются приборы для таких лабораторий с крупными стартовыми затратами. Неоправданно увеличивается персонал лаборатории, следовательно, увеличивается себестоимость элемент-определения. 2. Вся законодательная база в области аналитического контроля требует централизации контроля, а не децентрализации его по отдельным объектам анализа (водная, почвенная лаборатории и т.д.). Требуется концентрация аналитического контроля в госконтрольных органах и на крупных предприятиях-природопользователях. 3. Увеличение объема аналитических работ. Оно связано с объективной химизацией среды обитания. В случае экологических объектов требуется анализировать громадное число проб (из разных точек), обеспечивая представительность данных по каждому из множества показателей. Чтобы данные по разным точкам можно было сопоставлять, нужно получить их по надежным и однотипным (или даже унифицированным) методикам. Причем в каждой точке нужно отбирать все новые и новые пробы – ведь состав объекта (например, атмосферного воздуха) непрерывно меняется. Поэтому потребовались автоматизированные аналитические системы, оценивающие загрязнение водной или воздушной среды по данным множества станций и постов, где стоят автоматизированные анализаторы. Отсюда следует еще одна новая задача – существенное снижение трудозатрат на получение единичного результата. Эту задачу можно решить при использовании компьютеризированной аналитической техники, автоматов пробоподачи, эскалаторов подачи проб и других машиноуправляемых роботизированных устройств. Это позволяет одному оператору обслуживать до пяти разнотипных приборных комплексов. 4. Расширение номенклатуры объектов анализа и круга определяемых компонентов. К тому же резко увеличилось количество микро- и макрокомпонентов, присутствующих в пробах. Особую проблему составляет анализ свалок и отходов, нового объекта для аналитической химии. Экологический анализ всего того, что вытекает, испаряется из полигонов твердых бытовых отходов – это совершенно новая задача. В настоящее время по данным ВОЗ в промышленности используется 500 тыс. соединений (в основном органических), из которых более 40 тысяч является вредными для здоровья человека и около 12 тысяч токсичными. В России в почву вносят около 200 видов различных пестицидов, для 70% которых ПДК не определены. Из них только 10% являются нетоксичными. Т.е. системой нормирования охвачена только незначительная часть загрязнений, попадающих в окружающую среду. Многие соединения в результате взаимодействия с биотой превращаются в более токсичные. 5. Должна присутствовать определенная система наблюдений и выявления тенденций, на которую должны опираться диагноз и лечение болезни любого живого организма. Состояние окружающей среды также должно характеризоваться определенным набором параметров. Недостаточно просто установить превышение содержания ртути в воде, необходимо проследить тенденции его изменения в разный сезоны, при различных температурах и т.д. 6. Необходимость создания сети наблюдения за состоянием окружающей среды. В США наблюдение только за состоянием водных объектов ведется на 10000 станций.
ВОДА 1. Поверхностные природные воды. Природные воды классифицируют в соответствии с общей минерализацией: пресная вода – менее 1 г/кг; солоноватая – 1-25 г/кг; морская – 25-50 г/кг; рассолы (рапа) – более 50 г/кг. рН поверхностных вод изменяется в широком диапазоне от 4, 5 до 8, 5. Качественный состав матрицы природных вод характеризуется соотношением 6 главных ионов:
В зависимости от преобладания того или иного аниона воду принято называть: а) гидрокарбонатная и карбонатная; б) сульфатная; в) хлоридная. В свою очередь каждый класс делят на: – кальциевая; – магниевая; – натриевая. Кроме вышеуказанных основных макро-ионов в природных водах могут присутствовать: 1) растворенные газы О2, СО2, H2S, CH4 и др. 2) биогенные компоненты и элементы NH4+, NO2–, NO3–, Nорг, PO43– (рН> 9), HPO42–, H2PO4– (рН< 7), Pорг, SiO32–, Fе(II, III) и др. 3) микроэлементы. Сюда относят биометаллы Mn, Cu, Zn, Co и др.; неорганические природные загрязнители Ni, Cr, Cd, Pb, Hg, F– и др. В поверхностных природных водах микроэлементы входят в состав взвесей и коллоидов. В состав взвесей преимущественно входят катионы металлов, которые способны образовывать малорастворимые оксиды (MnO2), гидроксиды (Pb2+, Co2+, Ni2+, Cu2+). Амфотерные элементы, которые в природных водах содержатся в виде анионов (MoO42–), и анионы неметаллов сорбируются на взвесях хуже. При повышении рН сорбция микроэлементов возрастает. Формы существования различных элементов в природных водах зависит от рН, концентрации органических и неорганических лигандов в водах. 4) органические вещества: органический углерод, белки, амины, аминокислоты, карбоновые и оксикарбоновые кислоты, сложные эфиры, гумусовые и фульвокислоты, углеводороды, жиры, спирты, нефтепродукты, пестициды, СПАВ и др. 2. Подземные воды. На подземных горизонтах происходит насыщение воды минеральными компонентами и микроэлементами. Значительно увеличивается растворимость газов CO2, H2S, CH4. Растворенный кислород в подземных водах отсутствует Органические соединения могут присутствовать. 3. Морские и океанские воды. Химический состав воды морей и океанов имеет свои отличительные особенности. К таким особенностям можно отнести следующие: · огромное разнообразие качественного состава воды. Вода океанов аккумулирует разные по химическому составу воды со всей земной поверхности. · высокое солесодержание (в основном, NaCl). Общая минерализация морских и океанских вод достигает 35 г/кг. · постоянство во времени и однородность в разных частях океана основного химического состава воды. Это свойство обусловлено огромной массой Мирового океана, что создает стабильность солевой массы. Однородность обеспечивается постоянным водообменном между отдельными частями океана вследствие горизонтальных и вертикальных перемещений водных масс.
ВОЗДУХ И АТМОСФЕРНЫЕ ОСАДКИ Состав чистого сухого воздуха в объемных процентах следующий: N2 (78, 11), O2 (20, 95), Ar (0, 93), CO2 (0, 039), другие инертные газы (Ne, He, Kr, Xe) – 10–3-10–6, N2O (5·10–5), H2 (5·10–5), O3 ( 3·10–5), SO2, CH4, NO2 – 10–4-10–6. В воздухе присутствуют примеси газообразных веществ: – природного происхождения (SO2, NH3, HCl, H2S, CO, HF, CO2, CH4, молекулы органических веществ) – антропогенного происхождения (NO2, SO2, NH3, CO2, H2S, Cl2, Br2, HF, HBr, AsH3, PH3, галогенорганика, ароматические углеводороды, серусодержащая органика, пестициды и др.). В атмосфере содержатся не только газообразные вещества, но и большое количество твердых и жидких аэрозолей, пыль, дым, высокодисперсные агрегаты растворимых солей. При значительном загрязнении атмосферного воздуха рН осадков может снижаться до 4, 5-5 (кислотные дожди). ПОЧВЫ И ДОННЫЕ ОТЛОЖЕНИЯ Эти объекты могут содержать практически все элементы периодической системы Д.И. Менделеева. Особенности почв как объекта химического анализа: – большой набор элементов; – высокое содержание углерода и кремния; – большой диапазон концентраций, охватывающий 4-5 или даже 9-10 порядков; – профилированная дифференциация химического состава почв. Почва состоит из минеральной и органической частей. К органической части относят лигнин, флавоноиды и дубильные вещества, гумусовые кислоты, пигменты, липиды, углеводы, азотсодержащие соединения. Между двумя частями осуществляется постоянное органоминеральное взаимодействие. Химический состав почв зависит от их типа: тундровые, торфяные, подзолистые, серые лесные, пергнойно- карбонатные, черноземы, каштановые, бурые пустынно-степные песчаные, коричневые, бурые лесные, красноземы, сероземы. Средний элементный состав метрового слоя почвы по данным учебника Орлова Д.С., Садовниковой Л.К., Сухановой Н.И. (Химия почв. – М.: Высш. шк., 2005. – С. 45) составляет в %: O (49), H (0, 1), C гумуса (1, 4), С карбонатов (0, 24), N ~ (0, 1), P (0, 06), S (0, 09), Si (33), Al (6, 6), Fe (3, 2), Ti (0, 38), Mn (0, 16), Ca (1, 8), Mg (0, 9), K (1, 7), Na (1). Традиционный способ выражения химического состава почв – в виде массовой доли высших оксидов элементов, входящих в состав почвы. Например, минеральный макросостав дерново-подзолистой почвы в пересчете на абсолютно сухую навеску может быть представлен следующим образом (в %): SiO2 (73), Al2O3 (8, 7), Fe2O3 (2, 6), CaO (1), MgO (0, 7), K2O (2, 2), Na2O (1, 1). Недостающее до 100% количество приходится на MnO, P2O5, SO3 и на органические вещества. Органическая часть почвы состоит из живой органики (эдафон) и отмершей органики (гумус). К эдафону относят отмершие части живых организмов, не утративших своего анатомического строения. Гумификация эдафона ведет к образованию гумуса. Гумус – совокупность всех органических соединений, находящихся в почве, но не входящих в живые организмы или образования. Гумус состоит из специфических гуминовых веществ (темноокрашенные азотсодержащие высокомолекулярные соединения кислотной природы) и неспецифических соединений и промежуточных продуктов распада и гумификации. К последним относятся лигнин, целлюлоза, протеины, аминокислоты, аминосахариды, воска, жирные кислоты. Они присутствуют в почве в свободном состоянии или связаны с минеральными компонентами почвы. Гумусовые кислоты – главный и специфический продукты гумификации органических остатков в почвах. Это азотсодержащие высокомолекулярные оксикарбоновые кислоты с интенсивной темно- бурой или красновато-бурой окраской. Гумусовые кислоты (смесь) экстрагируют из почвы растворами щелочей, а затем по растворимости разделяют на гуминовые кислоты, гиматомелановые кислоты и фульвокислоты. Гуминовые кислоты – это группа веществ, извлекаемых из почвы щелочами в виде темно-окрашенного раствора (гуматов натрия, аммония, калия) и осаждаемых минеральными кислотами в виде аморфного осадка – геля при рН=1-2. В кислой среде гуминовые и гематомелановые кислоты выпадают в осадок. Состав гуминовых кислот (в %): С (46-62), N (3-6), H (3-5), O (32-38). Светлоокрашенные гумусовые вещества, остающиеся в растворе после подкисления щелочной вытяжки и отделения гуминовых кислот, называют фульвокислоты. Их состав (в %): С (36-44), N (3-4, 5), H (3-5), O (45-50). Фульвокислоты по сравнению с гуминовыми имеют более высокую растворимость и меньшую молекулярную массу. В составе молекул гуминовых и фульвокислот присутствуют различные функциональные группы: пептидные, амидные, альдегидные, карбоксильные, карбонильные, метоксигруппы, фенольные, хинонные. Минеральная основа д онных отложений речек, озер, водоемов аналогична почвам – это силикаты и алюмосиликаты. Более 90% донных отложений составляют материалы разрушения и эрозии горных пород на склонах водоемов и в руслах. Донные отложения вследствие их высокой сорбционной способности служат показателями антропогенного загрязнения водных экосистем и подлежат обязательному экологическому контролю.
Система химического анализа Рассмотрим конкретную экологическую аналитическую задачу. Предприятие собирается инвестировать средства в строительство уникального сооружения и нуждается в заключении о качестве почв того участка, где будет развернута стройка. Задача аналитиков – исследовать качество почвы в месте предполагаемого строительства. Совместно с заказчиком аналитик должен решить, какие компоненты следует определять в почве, какие общепризнанные надежные методики анализа следует для этого применить, в какой форме представить результаты анализа. Точная постановка аналитической задачи – необходимое условие того, что результаты анализа будут применены с пользой для дела. Процесс анализа начинается с превращения задачи в форме, поставленной потребителем, в собственно аналитическую задачу. Далее следует отобрать пробу из объекта исследования, т.е. отобрать пробу почвы. Затем следуют стадии пробоподготовки и измерения. Завершает процесс анализа обработка результатов, их сведение воедино, представление в отчете и передача потребителю. Круг замыкается и формируется так называемый аналитический цикл (рис. 1).
Рис. 1. Общая схема процесса анализа Аналитический цикл – это общая схема полного аналитического процесса. По определению Ю.А. Золотова метод – это определение принципов, положенных в основу анализа безотносительно к конкретному объекту и определяемому веществу. Методика – это полное описание всего хода анализа. В методике в форме подробных прописей оговариваются все детали анализа, включая отбор пробы и представление результатов. Представим на конкретном примере реализацию аналитического цикла. Перед аналитиком поставлена задача определения диэтиламина. Обычный алгоритм анализа меняется в зависимости от объекта анализа и матричных компонентов. Так, если в анализируемом образце присутствует только одно органическое вещество – диэтиламин, то наличие углерода является достаточным признаком специфичности. В этом случае задача решается хроматографическим методом с пламенно-ионизационным детектором. Если в объекте присутствуют другие органические вещества, но не амины, то специфической является –NH2 группа. В этом случае используют фотометрический детектор с β -динитростильбеном. Если в пробе есть первичные амины, то необходим метод детектирования именно вторичной аминогруппы =NH. Если есть другие вторичные амины, то надо дополнительно вводить признаки на наличие двух С2Н5-групп. Видно, что при определении одного вещества в различных композициях нужно применять разные методы, а при необходимости автоматизировать этот анализ – создавать разнообразные измерительные устройства в одном приборе. Один из главных принципов аналитической химии – селективность определения. Но в случае экоаналитического контроля это не всегда правильно. В объектах окружающей среды необходимо идентифицировать множество неизвестных компонентов, о присутствии которых там даже не подозревали. В данном случае нужна универсальная система химического анализа или многопараметрический анализ. Это, по сути, означает, что одновременно надо проводить качественный и количественный анализ. Для решения такой задачи необходимо изменить всю систему детектирования и использовать все признаки химических соединений. Такими признаками являются: 1) атомный состав. Методы определения элементного состава очень хорошо развиты. Пламенно-ионизационный детектор может показать к какому классу соединений (органические или неорганические) относятся вещества в пробе. 2) размер молекул. Это не селективный аналитический признак, но его используют в методах разделения смесей. Размер определяют с помощью молекулярных сит, полупроницаемых мембран. Возможно и прямое детектирование на основе селективной сорбции молекул определенного размера специфическими сорбентами с фиксированным размером микропор. 3) дипольный момент, характеризующий пространственное расположение эффективных зарядов в молекуле. Этот параметр может быть измерен с высокой точностью. 4) электронодонорные и электроноакцепторные свойства. 5) протонодонорные и протоноакцепторные свойства. Твердые электролиты, например, на основе фосфата титана меняют свои электрофизические свойства при контакте с протонодонорными или протоноакцепторными веществами. Возможны и фотоколориметрические детекторы с применением реагентов, образующих с определяемым компонентом окрашенный комплекс. 6) индекс хроматографического удерживания. Использовать этот индекс стало возможным после успехов в синтезе химически модифицированных сорбентов с заданными свойствами, позволяющими разделять вещества. 7) масса молекул. Знание массы позволяет однозначно определить брутто-формулу молекулы. В дальнейшем путем математического моделирования возможно записать все изомеры. В универсальной системе необходимо разделение функций измерения и функции обработки сигнала. Задача обработки сигналов детекторов решается с использованием ЭВМ, в которой предварительно сформирован банк химико-аналитических данных.
Атмосфера Для нормирования качества воздуха используют следующие нормативы. ПДКрз – предельно допустимая концентрация химического вещества в воздухе рабочей зоны в мг/м3. ПДКрз – это концентрация, которая при ежедневной (кроме выходных дней) работе в течение 8 часов на протяжении всего рабочего стажа не должна вызывать заболеваний или отклонения в состоянии здоровья в процессе работы или в отдаленные сроки жизни и жизни последующих поколений. ПДКмр – предельно допустимая максимальная разовая концентрация химического вещества в воздухе населенных мест в мг/м3. ПДКмр – концентрация вредного вещества в воздухе населенных мест, не вызывающая при вдыхании в течении 20 минут рефлекторных реакций в организме человека. ПДКсс – предельно допустимая среднесуточная концентрация химического вещества в воздухе населенных мест в мг/м3. ПДКсс – это концентрация, которая не должна оказывать на человека прямого или косвенного воздействия при неограниченно долгом (годы) вдыхании. Т.е. ПДКсс рассчитана на все группы населения и на долгий период воздействия и является самым жестким санитарно-гигиеническим нормативом. Интегральным показателем загрязнения атмосферы является индекс загрязнения атмосферы I:
где Сi – средняя концентрация i-того вещества; k – константа, зависящая от класса опасности загрязняющих веществ (табл. 3.1) Таблица 3.1. Классы опасности загрязняющих веществ воздуха
Гидросфера Для санитарной оценки воды водоемов хозяйственно-питьевого и рыбохозяйственного водоснабжения все используемые показатели могут быть разделены на 3 группы: – санитарные, куда входят микробиологические и паразитологические показатели (например, число микроорганизмов и число бактерий группы кишечных палочек в единице объема); – токсикологические, которые характеризуют безвредность химического состава воды; – органолептические, которые воспринимаются органами чувств человека (температура, прозрачность, цвет, запах, вкус). ПДКв – предельно допустимая концентрация химического вещества в воде водоемов хозяйственно-питьевого и культурного водопользования в мг/л или в мг/дм3. ПДКв – это концентрация, которая не должна оказывать прямого или косвенного влияния на организм человека в течение всей его жизни и на здоровье последующих поколений и не должна ухудшать гигиенические условия водопользования. ПДКвр (или ПДКрх) – предельно допустимая концентрация химического вещества в воде рыбохозяйственного водоема в мг/л или в мг/дм3. ПДКвр – это концентрация вредного вещества в воде, которая не оказывает вредного влияния на популяцию промысловых рыб. Сопоставим значения ПДКв и ПДКвр для некоторых токсикантов:
Видно, что для ряда соединений значение ПДКв значительно меньше, т.е. нормирование, например, акриламида в воде питьевого назначения значительно более жесткое по сравнению в водой для рыбохозяйственных целей. В то же время допустимое содержание тяжелых металлов в воде рыбохозяйственного назначения в несколько раз меньше. Это происходит не потому, что человек больше заботится о рыбах, а потому, что эти токсиканты обладают свойством аккумулироваться в рыбе, которую затем употребляет человек. Как и для воздуха существует интегральный показатель для оценки качества воды, который с одной стороны учитывает степень превышения ПДК отдельными токсикантами, а с другой стороны – частоту превышения. Зная концентрацию токсиканта в воде, рассчитывают баллы кратности Ki и баллы повторяемости Ni:
В монографии Б.Й. Набиванца с соавторами («Аналітична хімія поверхневих вод», издательство «Наукова думка», Киев, 2007 г.) состояние речных бассейнов предложено оценивать по величине общего экологического индекса IE, который рассчитывают как среднее арифметическое из трех индексов:
где I1 – индекс загрязнения компонентами солевого состава; I2 – трофо-сапробиологический (эколого-санитарный) индекс; I3 – индекс специфичных показателей токсичного действия. Индекс I3 позволяет оценивать экологическое состояние рек, в водах которых среди загрязняющих веществ преобладают компоненты с выраженным токсическим действием (тяжелые металлы, цианиды, нефтепродукты, фенолы, синтетические поверхностно-активные вещества). Присутствие этих веществ в поверхностных водах связано с хозяйственной деятельностью человека. Это бассейны Днепра, Днестра, Дуная, Южного Буга. Индекс I2 более информативен для описания состояния бассейнов рек, в которых основными загрязнителями вод являются трофо-сапробиологические (санитарно-экологические) показатели – кислород, минеральные формы азота и фосфора, взвешенные вещества, окисляемость воды (ХПК), БХП, рН, биомасса фитопланктона, численность сапрфитных бактерий. Такое загрязнение вод в основном связано с поступлением неочищенных либо недостаточно очищенных хозяйственно-бытовых стоков. Это бассейны Северского Донца, рек Приазовья, Западного Буга. Индекс I1 характеризует минерализацию воды, присутствие хлоридов, сульфатов.
Почва Для санитарной оценки почвы используют показатель ПДКп – предельно допустимая концентрация химического вещества в почве в мг/кг. ПДКп – это концентрация вредного вещества в пахотном слое почвы, которая не должна оказывать прямого или косвенного отрицательного влияния на соприкасающиеся с почвой среды и на здоровье человека, а также на самоочищающую способность почвы. Нормативы ПДК разработаны для веществ, которые могут мигрировать в атмосферный воздух или грунтовые воды. Интегральными показателями качества почв являются коэффициент концентрирования химического элемента в почве Кс и суммарный показатель загрязнения почвы Zc:
где Cф – фоновая концентрация элемента; КС – коэффициент концентрирования i-того элемента в пробе; n – число учитываемых элементов. Ориентировочная оценочная шкала опасности загрязнения почв по суммарному показателю выглядит следующим образом:
Продукты питания Для оценки безвредности продовольственного сырья и пищевых продуктов используют ПДКпр (ДУ) – предельно допустимые концентрации (допустимые уровни) токсичных элементов, соединений и ядохимикатов в продуктах в мг/кг. ПДКпр – концентрация вредного вещества в продукте питания, которая в течение неограниченно продолжительного времени (при ежедневном воздействии) не вызывает заболеваний или отклонений в состоянии здоровья населения. Попадающие в пищу из окружающей среды загрязнители делятся на две группы: 1) химической природы – токсичные металлы; – пестициды; – нитраты, нитриты, нитрозосоединения; – радионуклиды; – полициклические ароматические углеводороды; – диоксины; – гормональные препараты 2) биологической природы – микроорганизмы; – микотоксины; – антибиотики; – вирусы; – гельминты. Классы опасности токсикантам в этом случае присваивают в зависимости от их токсичности, которую характеризуют параметром ЛД50 (летальная доза, при которой гибнет 50% подопытных животных при введении токсиканта в желудок):
Как уже отмечалось выше, нормирование воздействия источника загрязнения на окружающую среду осуществляется с помощью показателей ПДВ и ПДС. ПДВ – масса вещества в отходящих газах источника загрязнения, максимально допустимая к выбросу в атмосферу в единицу времени (г/сутки). ПДС – масса вещества в сточных водах, максимально допустимая к отведению в данном пункте водного объекта в единицу времени с целью обеспечения норм качества воды (в мг/сутки). ПДС должен гарантировать достижение установленных норм при наихудших условиях для разбавления в водном объекте.
ВИДЫ ПРОБ Из материала курса «Аналитической химии» известно, что пробы различаются: – генеральная; – лабораторная; – аналитическая; – арбитражная. Однако при отборе проб объектов окружающей среды необходимо учитывать специфические требования: 1. Проба или серия проб, отобранных для анализа, должна быть представительной, т.е. характерной для данного природного объекта в месте их отбора. 2. Отбор проб, их транспортировка, сохранение и дальнейшая обработка не должны изменять содержание определяемых компонентов. 3. Объем или масса пробы должны полностью обеспечивать возможность выполнения всех запланированных аналитических определений. Виды проб при анализе объектов окружающей среды: 1. Простые или смешанные пробы. Простые пробы получают в результате одноразового отбора; они несут информацию о химическом составе воды, воздуха в определенном месте и в определенное время. Смешанные пробы – смесь простых проб, отобранных одновременно в разных местах или в одном месте за определенный промежуток времени. Эти пробы характеризуют средний химический состав воды или воздуха за определенный период. Смешанные пробы могут быть составлены непропорционально. Если установлено, что концентрация исследуемого компонента в средней части течения реки вдвое меньше, чем возле правого берега и в три раза меньше, чем возле левого берега, то смешанную пробу готовят сливанием объемов взятых в разных местах проб в соотношении 1 (середина): 0, 5 (возле правого берега): 0, 33 (возле левого берега). Смешанную пробу не рекомендуется отбирать за период больше одних суток. Ее нельзя отбирать для определения быстро изменяющихся показателей – растворенных газов, мутности, редокспотенциала. 2. Одноразовые (нерегулярные) или средние (регулярные) пробы. Одноразовый отбор используют при анализе глубинных подземных вод, химический состав которых постоянный, либо при контроле качестваводы природного объекта, для которого ранее были изучены закономерности изменения концентрации анализируемого компонента. Серийный отбор может производиться: – зональные пробы (пробы отбирают по схеме с разных глубин, в разных местах водного объекта); – через определенный промежуток времени (сезоны, декады, сутки, часы). Таким образом проверяют, как меняется качество воды со временем; – согласованные пробы. Они отбираются в разных местах по течению с учетом времени прохождения воды от одного пункта к другому. Такие пробы используют для оценки физических, физико-химических и биологических процессов, которые приводят к изменению химического состава воды вследствие самоочищения или самозагрязнения. ОТБОР ПРОБ ВОЗДУХА Основная погрешность при отборе проб воздуха связана с несоответствием состава пробы и состава анализируемой воздушной среды (отсутствие представительности пробы). Воздушная среда очень подвижна, а поступление вредных веществ может происходить как прерывисто, так и монотонно в зависимости от метеорологических и топографических факторов (направление и скорость ветра, температурные инверсии, атмосферное давление, влажность воздуха, рельеф местности, расстояние до источника загрязнения). Отбор проб воздуха осуществляют в двух режимах: непрерывном и разовом. В первом случае отбор проводят без перерывов в ходе всего времени наблюдения или в течение определенного времени; во втором – отбор в течение очень короткого промежутка времени. Наиболее достоверные данные, отражающие степень загрязнения воздуха газами и пылью, достигаются при непродолжительном отборе пробы. В этом случае фиксируются с достаточной точностью максимальные концентрации загрязнителей. Несмотря на то, что длительность отбора проб для большинства вредных веществ установлена 20-30 минут, согласно имеющимся наблюдениям, концентрация вредного вещества при такой экспозиции получается усредненной и в 3 раза ниже действительной максимальной, если пробы воздуха отбирать в течение 2-5 минут. Периодичность отбора зависит от характера технологического процесса, класса опасности, уровня загрязнения, времени пребывания обслуживающего персонала на рабочем месте. В зависимости от класса опасности вредного вещества отбор проводят не реже одного раза в 10 дней, в месяц или в квартал. Не существует универсального способа пробоотбора воздуха. В воздухе может содержаться до нескольких сотен токсичных компонентов в форме газов, паров, твердых частиц и аэрозолей, высокомолекулярных органических веществ, которые значительно отличаются по температуре кипения, молекулярной массе, сорбционным характеристикам.
КОНТЕЙНЕРЫ Используют для отбора органических и неорганических веществ в газообразном состоянии, летучих при обычной температуре веществ. Например, фреоны, хлоруглеводороды, бензол, толуол, углеводороды C2-C6, изопрен, CH4, CO, CO2, H2S, COS, CH3SH, CS2. Такой способ отбора используют для анализа газов и летучих веществ при обычной температуре. Он не связан с обогащением пробы анализируемыми компонентами. Его применяют для последующего газохроматографического анализа, иногда для спектрального анализа. Контейнеры – это сосуды различной формы. Контейнеры могут быть стеклянные, из нержавеющей стали и полимерной пленки. Типичный контейнер – стеклянная газовая пипетка, стеклянный шприц, стеклянные бутыли, резиновые камеры. Способы отбора газовых проб в контейнеры: 1. Пропускают анализируемый воздух через контейнер с небольшой скоростью. 2. Впускают воздух в предварительно вакуумированный сосуд. 3. Заполняют через ниппельное устройство. Примером простейшего контейнера является стеклянный газометр.
Рис. 1. Стеклянный газометр: 1– воронка; 2 и 3 – краны; 4 – баллон; 5 – пробка
Пробу набирают в предварительный вакуумированный сосуд. Перед работой газометр наполняют водой через воронку 1 при открытых кранах 2 и 3. Перед взятием пробы воздуха краны должны быть закрыты и газометр заполнен водой. Для взятия пробы трубку с краном 3 соединяют с местом отбора пробы, открывают кран 3 и вынимают пробку 5. По окончании пробоотбора закрывают кран 3 и пробку 5. Если должен быть известен объем взятого газа, то используют предварительно калиброванный газометр. Из взятой пробы отбирают аналитическую пробу газа: для этого трубку с краном 3 газометра присоединяют к газоанализатору и открывают краны 2 и 3. Вода из воронки стекает вниз и вытесняет пробу в газоанализатор. Отбор небольших проб газа Для отбора небольших объемов проб газа (250-500 мл) используют трубку с кранами (газовую пипетку), которую присоединяют к водоструйному насосу. Через эту трубку некоторое время «протягивают» испытуемый газ, затем краны закрывают (рис. 3.2). Для переведения пробы из пипетки в газоанализатор последний присоединяют к верхнему концу пипетки, другой конец пипетки соединяют с сосудом, содержащим жидкость, в которой газ не растворяется (вода, насыщенный раствором NaCI, минеральное масло, ртуть) и вытесняют газ в газоанализатор.
Рис. 2. Пипетка и насос для засасывания газа
При другом способе пипетку одним концом присоединяют к отборной трубке, другим – к склянке с жидкостью (рис. 3).
Рис. 3. Пипетка для отбора пробы газа
При открытых кранах склянку поднимают, и жидкость заполняет пипетку до верхнего крана. Верхнюю часть пипетки соединяют с газоподводящей трубкой. Затем склянку опускают, и газ входит в пипетку. Первые 3-4 порции газа выпускают из пипетки в атмосферу и набирают новую порцию, опуская жидкость до нижнего крана; после этого закрывают краны. Поднимая склянку, переводят пробу в газоанализатор. Отбор проб в эвакуированные сосуды (для ядовитых проб) Приемники для отбора пробы газа, которые называют эвакуированные сосуды, представляют собой баллоны вместимостью от 0, 5 до 3 л, иногда даже 10 л. (рис.4).
а б в Рис. 4. Эвакуированные сосуды: а – колба с краном; б – склянка; в – баллон
Баллон снабжен краном. Перед отбором пробы из баллона насосом откачивают воздух до 40 мм Hg (≈ 5, 3 кПа), кран закрывают, баллон взвешивают и переносят к месту отбора пробы. Трубку баллона присоединяют к газоотборной трубке и кран открывают. После отбора газа кран закрывают и баллон вновь взвешивают; по разности находят массу взятой пробы. Эвакуированные сосуды применяют при анализе ядовитых газов или паров в воздухе производственных помещений. Для отбора проб воздуха распространены мешки из полимерных пленок (рис. 5). Материал мешков – пленка поливинилфторидная (ПВФ), поливинилхлоридная (ПВХ) с мембраной из силикона или тефлона. Сбоку расположен клапан. Отбор пробы воздуха из пленки производится шприцем, которым прокалывают полимерную прокладку (мембрану из тефлона или силикона). Объем мешков может составлять от 0, 5 до 96 л (есть и 170 л). Заполняют мешки из полимерной пленки анализируемым воздухом через клапан (муфту) ниппельного типа.
Рис. 5. Различные типы полимерных мешков для отбора проб воздуха
Побочные процессы при отборе проб газов: 1. Сорбция (хемосорбция) определяемых компонентов может составлять до 40-70%. Для снижения сорбции в стеклянных сосудах их обрабатывают парами анализируемых соединений, этим можно сократить потери на 50%. Тефлон при комнатной температуре адсорбирует до 40% бензола. 2. Химические реакции компонентов пробы между собой и с материалом контейнера в присутствии влаги, света и кислорода воздуха. Для их снижения надежней использовать мешки из тефлона. 3. Потери части вещества из-за негерметичности контейнера и проницаемости полимерной пленки. Например, через тефлоновую пленку CH4 и C2H2 легко диффундируют. Отбор проб воздуха в канистры имеет следующие преимущества: 1. Возможность получения представительной пробы. 2. Интегрирование ЛОС за определенный промежуток времени. 3. Облегчение транспортировки отобранных проб (идентичность состава пробы не нарушается в течение недели). 4. Достигаются хорошие точность и воспроизводимость. Канистры из нержавеющей стали предварительно подвергают процессу электропассивации, чтобы снизить количество активных полярных мест. Кроме того канистры предварительно очищают: проводят серию процессов «вакуумирование – заполнение» с помощью не масляного компрессора и с использованием ультрачистого азота. Остаточное количество ЛОС после этого не превышает 0, 01-0, 1 ppb. При пробоотборе канистру вакуумируют до 50 мм Hg, а затем заполняют воздухом через вентиль тонкой регулировки (время отбора от 5 минут до 25 часов).
Моря и океаны. Выбор места отбора должен проводиться с учетом приливных течений. Удаленность от берега, направление ветра, плотность воды, состояние дна, судоходство могут повлиять на химический состав пробы. Следует учитывать возможность стоков или выбросов в месте отбора проб. Реки и ручьи. Пробы отбирают в местах наиболее быстрого течения в фарватере. Если вода 2х речек смешивается или смешивается речная и сточная вода, то пробу надо отбирать в местах полного перемешивания водных масс. Глубина отбора, как правило, 20-30 см. Сточные воды. Они отличаются непостоянным составом. Место отбора проб сточных вод выбирают после ознакомления с технологией производства. Пробу отбирают в турбулентных, хорошо перемешанных потоках на прямолинейных участках водоотводящих устройств. Если сточные воды отводятся в водный объект, то пробы отбирают у их выпуска в водоем. Частота отбора проб определяется с учетом реального расхода и состава сточных вод данного производства. В аварийных ситуациях, при ремонте очистных сооружений частоту отбора увеличивают. Питьевая вода. Пробу отбирают перед поступлением в распределительную сеть, а также в самой сети после спуска воды в течение не менее 15 мин при полностью открытом кране. Частота отбора проб: на водопроводах споверхностным источником водоснабжения – 1 раз в неделю или ежесуточно в зависимости от численности населения. Чем больше численность населения, тем чаще пробоотбор. На водопроводах с подземным источником отбор проб проводят от 1 раза в неделю до 1 раза в месяц. Объем отбираемых проб зависит от числа определяемых компонентов: – неполный анализ (соответствие гигиеническим 1 л нормативам, некоторые контрольные определения) – более подробный анализ 2 л – полный анализ или определение компонентов с 10 л очень низким содержанием
Приспособления для отбора проб воды называются батометры. Они различны по конструкции, начиная от обычной молочной бутыли и заканчивая автоматическими пробоотборниками вместимостью 1-4 л, массой 2, 5-30 кг. Среди современных батометров выделяют: Сериальные – имеющие боковой подвес, позволяющий легко укрепить их на тросе или снять с троса, вытравленного за борт и натянутого грузом нижних батометров. Малые – компактной конструкции. Первый малый батометр был предложен в 1935 году и изготовлен для дрейфующей станции «Северный полюс». Концевые – применяются, чтобы избавиться от подъѐ ма и спуска излишней тяжести груза при спуске нескольких батометров. Проба воды из концевого батометра, в котором используется стеклянный или другой химически стойкий сосуд, более надѐ жна (является контрольной). Вес батометра Международной гидрографической лаборатории 12 кг, полная ѐ мкость 1700 см³, ѐ мкость внутреннего сосуда 450 см³. Донные – предназначены для получения проб воды из слоя, непосредственно прилегающего ко дну. Строятся как одиночные (по типу концевых) и закрываются автоматически от прикосновения ко дну (без посыльного груза). Первый донный батометр создан в 1870 году. Промерные – употребляются для добывания придонных проб воды во время глубоководного промера. Такой батометр лѐ гкий (около 3 кг) и закрывается автоматически при начале подъѐ ма лотлиня (проволоки). Специального назначения – применяются, когда необходимо с одной глубины получить большое количество воды (для полного химического анализа, гидробиологических работ), имеют большой объѐ м (10 литров и более). Батометры также можно классифицировать и по другим признакам. С учѐ том потребности во времени наполнения батометры могут быть быстрого (мгновенного) или длительного наполнения объѐ ма. Батометры также можно классифицировать и по другим признакам. С учѐ том потребности во времени наполнения батометры могут быть быстрого (мгновенного) или длительного наполнения объѐ ма. Батометр быстрого (мгновенного) наполнения имеет крышку, которая закрывается на заданной глубине в результате переворачивания батометра, происходящего под воздействием посылаемого по тросу груза. Одновременно установленным на батометре термометром регистрируется и температура воды. Сходное устройство имеет речной батометр Жуковского, но он опускается в водоѐ м в горизонтальном положении. В батометр длительного наполнения вода поступает со скоростью течения воды в исследуемой точке. Рис. 3.10. Батометр Руттнера Рис. 3.11. Батометр Молчанова ГР-18
Предназначен для взятия проб воды с различных глубин водоемов, с одновременным измерением температуры воды исследуемого слоя при температуре окружающей среды от +1 до +40°С.
Рис. 3.12. Морской батометр БМ-48 (батометр Ф. Нансена)
Морские батометры имеют длину 65 см, вес 4, 3 кг, ѐ мкость 1 л. Батометр состоит из латунного цилиндра, окрашенного в белый или серый цвет. На обоих концах цилиндра имеются крановые затворы со щелевидными отверстиями длиной около 60 и шириной 12 мм. Трение кранов регулируется спиральными бронзовыми пружинами, которые прижимаются к кранам гайками. К расширенным концам обоих кранов прикреплены два параллельных рычага, посредством которых краны закрываются и открываются. Концы рычагов соединены на шарнирах со штоком, этим достигается одновременность действия обоих кранов. Для предохранения запорных кранов от самопроизвольного открывания на штоке имеется конусный выступ, который при опрокидывании батометра заскакивает за прикреплѐ нную к батометру пластинку и тем самым прочно удерживает краны в закрытом состоянии. Прибор крепится к тросу с помощью зажимного устройства на нижнем конце батометра и спускового устройства на верхнем конце. Для подвешивания и сбрасывания посыльного груза на расположенный ниже батометр на зажиме смонтировано срабатывающее устройство. Для увеличения опрокидывающей силы к спусковому устройству прикреплена направляющая пластина. Для извлечения проб воды в верхней части батометра имеется сливной кран, а в нижней – воздушный клапан, который при выливании воды приоткрывается для доступа воздуха в батометр. К батометру прикреплены два угольника, на которых укрепляется оправа для глубоководных термометров. Рис. 3.13. Батометр Нискина (Niskin bottle)
Этим батометрам не нужно переворачиваться, чтобы захлопнуться. Иногда их вешают на трос по одному, но чаще всего их собирают в розетту (rosette) и опускают в воду. При достижении определѐ нной глубины батометры закрываются. Потом на палубе пробы из батометров отбираются для анализа. Рис. 3.14. Отбор проб морской воды с помощью батометров
В простейшем случае можно использовать стеклянные, фарфоровые или пластмассовые сосуды с притертыми или завинчивающимися пробками (герметичная укупорка). Материал сосуда зависит от примесей. Питьевую воду отбирают в сосуды из стекла, полиэтилена. Пробы, в которых необходимо определять органические вещества, только в стеклянные сосуды. Очень важна чистота сосуда и пробки. Стеклянную посуду тщательно моют и обеззараживают, отмывают от кислот, пропаривают водяным паром. Полиэтиленовые сосуды ополаскивают ацетоном, HCl (1: 1), дистиллированной водой. Вымытую посуду ополаскивают анализируемой водой. Корковые пробки кипятят для очистки в дистиллированной воде, резиновые – в 5% растворе HCl, затем в 20% растворе NaOH, отмывают дистиллированной водой и хранят в стеклянных банках с крышками. К каждой пробе составляется сопроводительный документ, который должен содержать сведения: – номер бутыли (тары); – наименование вида вод; – место отбора пробы; – дата и время отбора; – способ отбора (тип пробоотборника, приспособления); – вид пробы (простая, смешанная); – периодичность отбора пробы; – сведения о консервировании пробы или обеспечении ее сохранности; – ФИО и подпись лица, участвующего в отборе проб и их подготовке.
ОТЛОЖЕНИЙ, ШЛАМОВ При отборе проб почвы следует учитывать адсорбционную способность почв, протекающие в них сложные физико-химические, биологические процессы, неоднородность распределения токсикантов в почвах. Перечислим некоторые особенности пробоотбора и пробоподготовки почв: 1. Максимальное содержание металлов в почвах (Cd, Pb, Ni, Zn, Hg, Cu, Fe, Mo, Sn) наблюдается на расстоянии 1-5 км от источников загрязнения. На расстоянии 15-20 км содержание металлов в почве соответствует фоновому уровню. 2. Глубина проникновения тяжелых металлов в почву не превышает 20 см. При сильном загрязнении – до 160 см. Для почв вне зоны источника загрязнения, распределение тяжелых металлов равномерное. 3. Наибольшей миграционной способностью обладают Hg и Zn, которые как правило равномерно распределяются в слое почвы 0-20 см. 4. Свинец чаще накапливается в поверхностном слое 0-2, 5 см, кадмий занимает промежуточное положение. Все это приводит к неравномерному извлечению загрязнений различной природы. 5. Загрязнение почвы фтором происходит от предприятий по переработке фторсодержащего сырья (суперфосфатные и кирпичные заводы, химические, стекольные заводы, алюминиевые комбинаты). Фтор мигрирует в растения и распространяется на очень широких площадях, шире, чем зона влияния предприятия. На растения воздействуют и газообразные фториды. 6. Почва прекрасный адсорбент пестицидов. Механизм перераспределения пестицидов в почвах до настоящего времени практически не изучен ни для одного вещества. 7. При пробоподготовке проб почв существует опасность их загрязнения растворителем или экстрагентом. Кроме того, растворитель будет извлекать лишь растворимые вещества. Поэтому вытяжку надо делать водой, органическими полярным и органическими неполярными растворителями. Отбор проб почв проводят в соответствии со стандартом ГОСТ 17.4.4.02.84. Закладывают пробные площадки – часть исследуемой территории, характеризующаяся сходными условиями. При общем загрязнении почв пробные площадки намечают по координатной сетке. При локальном загрязнении для пробных площадок применяют систему концентрических окружностей вокруг источника загрязнения, указывают номер окружности и азимут места отбора проб. Размер пробной площадки, количество и вид пробы различаются в зависимости от цели исследования. Обычно закладывают 1 или 2-3 площадки размером 25 м2 каждая. С каждой площадки отбирают по 5 точечных проб. Отбор проводят с 3-х разных глубин (горизонтов) в зависимости от поставленной цели (определение степени загрязнения поверхностного слоя, миграция химического вещества по профилю почвы и т.д.): 0-20 см; 20-40 см и 40-60 см. Объединенную пробу готовят методом квартования из смеси не менее 2 точечных проб, отобранных с разных слоев, массой не менее 1 кг. Если обследуемое поле (участок) расположено на различных элементах рельефа (плато, склон, подножье склона), то объединенная проба почвы отбирается с каждого элемента рельефа. При контроле загрязнения почв промышленными источниками площадки для отбора проб располагают на площади трехкратной величины санитарно-защитной зоны вдоль векторов розы ветров на расстоянии 100, 200, 300, 500, 1000, 2000, 5000 м и более от источника загрязнения. При контроле почв в районе точечных источников загрязнения (выгребные ямы, мусоросборники и т.п.) пробные площадки размером не более 5 х 5 м закладываются на разном расстоянии от источника и в относительно чистом месте (контроль). При изучении загрязнения почв транспортными магистралями пробные площадки закладываются на придорожных полосах с учетом рельефа местности, растительного покрова, метео- и гидрологических условий. Пробы почвы отбирают с узких полос длиной 200-500 м на расстоянии 0-10, 10-50, 50-100 м от полотна дороги. Одна смешанная проба составляется из 20-25 точечных, отобранных с глубины 0-10 см. При оценке почв сельскохозяйственных территорий пробы почвы отбирают 2 раза в год (весна, осень) с глубины 0-25 см. На каждые 0-15 га закладывается не менее одной площадки размером 100-200 м2 в зависимости от рельефа местности и условий землепользования. Инструментами для отбора проб служат: 1. почвенный бур, который позволяет отбирать пробу с глубины до 2-х метров (рис. 3.20); 2. перфораторы (до 10 м); 3. лопата (20-25 см). Рис. 3.20. Буры для отбора почв:
Отобранные пробы нумеруют и регистрируют в журнале с указанием порядкового номера, числа, горизонта и глубины взятия пробы, рельефа местности, типа почвы, целевого назначения, даты отбора. На этикетке также указывается ФИО отборщика. Для отбора речных и озерных отложений используются устройства типа земленасосного снаряда или землечерпалки, устройство которых представлено на рис. 3.21. Рис. 3.21. Землечерпалки типа Ponor (а) и Ekman (б)
От грязей или шламов отбирают либо единичные, либо комбинированные пробы. Если грязь, шлам содержат большое количество жидкости и они однородны, то используют стандартные отборники, применяемые для воды. Для отбора материалов с высоким содержанием твердой фазы используют отборник типа Rawson, который по существу представляет собой отсасывающее устройство (рис. 3, 22, а) Рис. 3.22. Пробоотборники: а – типа Rawson (положения 1, 2); б – типа Mason; в – типа Lenz (1 – вид сбоку, 2 – вид спереди)
Отсос обеспечивается резиновой грушей, которая сдавливается металлическими пластинами, открывая доступ для осадка грязи. Устройство выносится на поверхность, и из груши извлекается захваченный материал. Полужидкие грязи или шламы обычно отбирают с помощью драг, используемых при отборе проб донных отложений. На рис. 3.22, б изображена драга типа Mason. Применяют также отборники плунжерного типа. Схематичное изображение подобных устройств представлено на рис. 3.22, в. МЕТОДЫ ИЗВЛЕЧЕНИЯ ЗАГРЯЗНЯЮЩИХ ВЕЩЕСТВ (ПРОБОПОДГОТОВКА) Абсорбционное улавливание Абсорбционное улавливание загрязнений воздуха – это отбор веществ, находящихся в воздухе в газо- и парообразном состоянии, в жидкие поглотительные среды (вода, кислоты, спирты, органические растворители), в которых определяемое вещество растворяется или химически связывается поглотительной средой. Абсорбционное улавливание дает возможность одновременно концентрировать все определяемые примеси кроме аэрозолей и твердых частиц. Для абсорбционного улавливания токсикантов загрязненный воздух пропускают через поглотительную склянку ( абсорбер), содержащую несколько миллилитров растворителя, природа которого определяется составом пробы. На рис. 3.6. изображены абсорбер широко используемые в практике санитарно-химического анализа.
1 2 3 4 Рис. 4.6. Поглотительные приборы (абсорберы) из стекла: Зайцева (1), Киселева (2), Рыхтера (3 – малый, 4 – скоростной)
Наибольшее распространение получили абсорберы со стеклянными пористыми пластинками, поглотительные сосуды Рыхтера и Зайцева. Пористая пластинка уменьшает размер пузырьков воздуха и увеличивается контакт воздуха с раствором. Полнота поглощения зависит от природы анализируемых примесей и абсорбента, их концентрации, скорости потока воздуха. Для летучих органических поглотителей абсорбер охлаждают смесью льда и соли. Особенно эффективным является поглощение, основанное на химических реакциях абсорбируемых веществ с поглотителями. Например, реакционно способный триоксид серы поглощается раствором хлорида бария, а диоксид углерода – раствором гидроксида кальция: SO3 + H2O = H2SO4 H2SO4 + BaCl2 = BaSO4 + 2HCl CO2 + H2O = H2CO3 H2CO3 + Ca(OH)2 = CaCO3 + 2H2O Причины возможных погрешностей при аспирационном улавливании: 1. Неправильное измерение объема аспирируемого воздуха. 2. Пренебрежение агрегатным состоянием анализируемых веществ. 3. Не оптимальный выбор поглотительных сред и скорости аспирации воздуха. 4. Наличие микропримесей сопутствующих или посторонних веществ в поглотительных растворах. Вода, органические растворители могут содержать примеси. Например, примесь воды в поглотительной нитросмеси при определении бензола в воздухе способствует неполной реакции нитрования и уменьшению количества динитробензола, по количеству которого судят о содержании бензола в воздухе. Характеристика фотометрических методов определения некоторых токсичных газов в воздухе после их абсорбционного улавливания представлена в табл. 3.3. Абсорбционный раствор можно затем использовать для повторного концентрирования некоторых токсикантов, т.к. даже хроматографическим методом с высокочувствительным пламенно-ионизационным детектором и детектором электронного захвата не всегда удается достичь необходимого предела обнаружения токсикантов. Еще один важный прием в пробоподготовке после абсорбции токсикантов – дериватизация. Дериватизация – это получение производных определяемых компонентов. Она особенно эффективна при использовании хроматографических методов, т.к. использование специфических реагентов, которые вступают в реакцию только с контролируемыми компонентами, значительно повышает надежность идентификации целевых компонентов. Например, формальдегид является одним из важнейших загрязнителей воздуха. При аспирационном улавливании воздух пропускают через абсорбер, содержащий реагент ацетилацетон в ацетатном буферном растворе. В результате реакции между формальдегидом и ацетилацетоном образуется нелетучее производное формальдегида, окрашенное в желтый цвет. Поглотительный раствор нагревают на водяной бане и измеряют оптическую плотность окрашенного раствора. Примером дериватизации в газовой хроматографии может служить до 2·10–7% HCN в воздухе. Пары кислоты поглощают водным раствором щелочи. В щелочной среде происходит реакция цианид-иона с пентафторбензилбромидом с образованием пентафторбензилцианида: Образование фторпроизводного HCN позволило использовать селективный к галогенам детектор электронного захвата.
Сорбция (адсорбция) Сорбционное извлечение примесей токсичных веществ из загрязненного воздуха является главным и широко применяемым способом пробоотбора. Способ универсальный. Из воздуха извлекается весь спектр загрязнителей – от газов до высококипящих органических соединений (кроме твердых частиц и аэрозолей). Воздух с помощью различных аспирационных устройств пропускают через трубку с сорбентом, а после завершения пробоотбора транспортируют ее в лабораторию, где сконцентрированные примеси извлекают (термодесорбция, экстракция) и анализируют подходящим методом (хроматография, спектральный анализ, электрохимические методы и т.д.). Типичными трубками (ловушками) с сорбентами являются, например, трубки с активным углем – наиболее дешевые и эффективные пробоотборные устройства (рис. 3.7).
Рис. 3.7. Ловушка-концентратор для извлечения примесей из воздуха: 1 – заглушки из пластика; 2 – стеклянная трубка с оттянутым концом; 3 – высокочистое стекловолокно; 4 – сепаратор из пенопласта; 5 – пружинный запор для фиксирования слоя угля; 6 – основной слой активного угля (100 мг) с точно известной удельной поверхностью и размером частиц; 7 – резервный слой угля (50 мг); 8 – предохранитель, позволяющий при необходимости легко отломать кончик трубки Активный уголь позволяет извлекать из воздуха большинство известных органических соединений. Однако уголь хорошо сорбирует влагу и термодесорбция многих ЛОС с углей затруднена. В качестве сорбентов для заполнения концентрационных трубок используют также: 1) активный уголь (кокосовый, нефтяной, древесный); 2) SiO2∙ nH2O; 3) Al2O3; 4) пористые полимеры (тенакс, хромосорб, порапак и др.); 5) графитированные сажи и углеродсодержащие полимеры (карбосив, карбопак и др.); 6) молекулярные сита. Улавливание из воздуха аэрозолей имеет свои специфические особенности. Попадающие в атмосферный воздух твердые частицы (пыль, сажа) или аэрозоли (пестициды, ПАУ, металла, неорганические соли и др.) значительно превышают размеры атомов и молекул и не улавливаются обычными сорбентами. Аэрозоли улавливают на фильтры. Они задерживают частицы размером 0, 1-0, 2 мкм. Фильтры могут быть: – из стекловолокна; – керамики; – полимерных материалов (например, фильтры АФА-ВП изготовлены из тонковолокнистого перхлорвинилового волокна). После отбора пробы фильтры растворяют: АФА-ХА (ацетилцеллюлоза) – в смеси кислот HNO3 + HClO4; АФА-ХП (перхлорвинил) – в кислоте или в ацетоне, дихлорэтане. АФА-ХС (полистирол) – в щелочи. Отбор проб на фильтры используется для последующего гравиметрического, полярографического, рентгеновского и эмиссионного спектрального методов анализа токсикантов. Фильтр закрепляют в фильтродержателе (рис. 3.8, 3.9).
Рис. 3.8. Фильтродержатели для фильтров (аллонжи)
Фильтродержатели для фильтров (аллонжи), аэрозольные патроны изготавливаются из алюминия или ударопрочного полистирола. Цифры в маркировке указывают на размеры используемого фильтра. Например, фильтродержатель ИРА-10-2М применяется в комплекте с фильтрами АФА-10
Рис. 3.9. Аналитические аэрозольные фильтры
Аналитические аэрозольные фильтры АФА-ВП, АФА-РМ, АФА-Х, АФА-РСП (в фильтродержателях) изготавливаются в нескольких исполнениях в зависимости от площади рабочей поверхности (3 см, 10 см, 20 см, 40 см) и материала фильтрующего элемента. Стандартная методика атомно-абсорбционного определения неорганических соединений ртути основана на предварительном концентрировании солей и оксидов ртути при пропускании воздуха через перхлорвиниловый фильтр АФА-ХА-20 в течение 5 минут. Затем фильтр кипятят в растворе KMnO4 + H2SO4 и полученный раствор вводят в атомизатор. Улавливание аэрозолей на фильтры было использовано для определения 65 элементов (в основном металлов) атомно-эмиссионным методом с индуктивно связанной плазмой в атмосфере. Для анализа городских аэрозолей использовали метод рентгеновской флуоресценции после улавливания аэрозолей на мембранном фильтре на основе ацетата целлюлозы. Полициклические ароматические хиноны улавливают на фильтре из атмосферных аэрозолей, извлекают хиноны с фильтра экстракцией органическими растворителями и определяли методом высокоэффективной жидкостной хроматографии. Перечень примеров можно продолжить.
ВЕЩЕСТВ ИЗ ВОДЫ Твердофазная экстракция Это сочетание сорбционных и ионообменных процессов (жидкостно- твердофазное разделение). Твердофазная экстракция гораздо более быстрый способ по сравнению с классическими методами выделения. Способ пригоден для извлечения из воды мало-, средне-, и высокополярных загрязнителей. Экстракция может быть проведена на картридже (патроне, заполненном сорбентом) (рис. 3.17), либо на мембранных дисках. Затем проходит элюирование малым объемом элюента или термодесорбция. В качестве сорбентов используют: полимерные сорбенты (амберлиты, тенакс, хромосорб, порапак и др.), активный уголь, графитированные сажи, синтетические иониты. Порошкообразную твердую фазу обычно помещают в маленький патрончик наподобие пластмассового шприца. Сверху наливают раствор пробы и пропускают его через слой сорбента при помощи поршня (под действием избыточного давления) или центрифугирования. При этом происходит извлечение следовых количеств органических веществ, их концентрирование на колонке и отделение от матрицы пробы. Патрончики для твердофазной экстракции применяют для выделения физиологически активных веществ (лекарственные препараты, наркотики) из биологических проб. В этих случаях для повышения производительности их обычно используют сериями, по 12 или 24 штук одновременно, в сочетании с вакуумным насосом и автоматизированными жидкостными системами (рис. 3.18). В автоматизированных приборах для обработки большого числа проб (например, рис. 3.19) широко применяются 96-луночные планшеты с маленькими лунками (так называемые микролитровые планшеты).
Мембранные методы Использование полимерных проницаемых мембран – это наиболее перспективный метод извлечения и концентрирования загрязняющих веществ при анализе больших проб воды. Мембраны чаще всего изготавливаются из ацетилцеллюлозы, полиамида, полифуранов, полиакрилонитрилов, полиэтилена и т.д. Мембранный диск помещают в картридж. Пробу воды под вакуумом пропускают через мембрану и при этом происходит фракционирование определяемых веществ. Сконцентрированные на дисках и патронах загрязняющие вещества затем обычно элюируют органическими растворителями. Последующие методы анализа – атомно-абсорбционная, атомно-эмиссионная с индуктивно связанной плазмой спектроскопия, ВЭЖХ.
Газовая экстракция Продувку проб с последующим улавливанием используют при определении в воде газов и легколетучих органических соединений (ЛОС). Через пробу воды продувают инертный газ, он захватывает ЛОС, которые затем улавливают на адсорбентах (например, на активный уголь) и конденсируют в криогенной ловушке. Ловушка с адсорбентом встроена в десорбционную камеру с нагревателем. Устройство для стриппинга монтируется сразу на газовом хроматографе с последовательно подключенным детектором электронного захвата или пламенно- ионизационным детектором. Иногда устройство для газовой экстракции делают по методу замкнутой петли, т.е. газ циркулирует через нагретую пробу воды и через ловушку с активным углем в течение 90 минут. Сконцентрированные на угле летучие вещества затем экстрагируют CS2 или хлористым метиленом.
Спрэй-экстракция Это сравнительно новая технология пробоподготовки, которую используют для извлечения и концентрирования ЛОС. По сути, это вариант газовой экстракции. Водную пробу под давлением инертного газа разбрызгивают через узкое сопло, в результате чего образуются очень мелкие капли водного раствора в экстракционной камере. Большая общая площадь поверхности между жидкостью и газом обеспечивает быстрое установление равновесия. Поток газа с извлеченными из капелек жидкости аналитами направляется в трубку с тенаксом, в которой концентрируются извлеченные из воды ЛОС. Далее – термодесорбция и определение целевых компонентов. Микроволновая пробоподготовка Воздействие физических полей на химические процессы, в частности, микроволн, позволяет интенсифицировать ряд стадий при выполнении экологических анализов. 1. Микроволновое излучение способствует ускорению разложения проб, в том числе при повышенных температурах и давлении. 2. Происходит ускорение собственно аналитических реакций, что позволяет интенсифицировать предварительную подготовку аналитических форм, способных к последующему взаимодействию. Это особенно касается Pt-металлов, которые образуют кинетически инертные комплексы. 3. Интенсификация процессов приготовления сорбентов, экстрагентов, аналитических реагентов. Например: микроволновая обработка значительно ускоряет сушку геля для приготовления тест- методов на основе золь-гель технологий. 4. В экологической аналитической химии микроволновое воздействие увеличивает эффективность извлечения примесей токсичных компонентов из матрицы, способствует более полной десорбции токсикантов из сорбционных пробоотборных трубок, увеличивает степень извлечения загрязнителей из картриджей при анализе воды, летучих и малолетучих токсикантов из почвы, донных осадков, твердых отходов, из биологических тканей. Например, определение тяжелых металлов в сточных водах крупного промышленного региона осложняется тем, что стоки представляют из себя сложный и малопредсказуемый объект анализа. Металлы в такой воде присутствуют в растворенной форме, во взвешенном состоянии, кислотоэкстрагируемые и общие (валовое содержание).
Жидкостная экстракция В отличие от термодесорбции здесь нет опасности термического разложения компонентов пробы. Жидкостная экстракция используется для извлечения малолетучих и нелетучих соединений из почв, донных осадков, твердых бытовых и химических отходов, пластмасс. Экстракцию используют, например, при извлечении из почвы взрывчатых и отравляющих веществ в местах обезвреживания снарядов. Экстрагировали ТНТ, люизит (2 хлормышьяковая кислота), алкилфосфоновые кислоты (отравляющие вещества нервно-паралитического действия), зарин, зоман. Основной метод выделения пестицидов и полихлорбифенилов из почв – тоже экстракция. Для экстракции используют экстрактор Сокслета (рис. 3.23).
Рис. 3.23. Экстрактор Сокслета
Экстрактор Сокслета используют для экстракции необходимых компонентов не только из почв, но и из растительного сырья и т.д. При экстракции в колбу на 0, 5-1 л (B) наливают 100 мл растворителя легче воды (пентан, диэтиловый эфир, петролейный эфир, гексан). В колбу C помещают пробу, упакованную в марлевый мешочек. Растворитель поддерживают в кипящем состоянии. Через трубку D пары растворителя поступают в холодильник A, где конденсируются и падают по каплям сверху на пробу. По мере подъема уровня растворителя он насыщается извлекаемыми компонентами. После того, как уровень растворителя достигнет верхнего уровня сифона E, он сливается через него в колбу B и, продолжая кипение, вновь начинает поступать в экстрактор C. Процесс экстракции может продолжать сколь угодно долго. После окончания экстракции растворитель (а точнее сказать, экстракт) сливают из колбы B в подходящую емкость и используют для дальнейшего анализа. Однако органические растворители токсичны, дороги. Процесс извлечения длительный, экстракция в аппарате Сокслета может длиться от 8 до 40 часов.
Парофазный анализ Пробу (например, исследуемой воды) помещают в специальныйисосуд, плотно закрывают и термостатируют для перевода летучих компонентов в газовую фазу. После установления равновесия между газовой и жидкой фазой аликвоту газовой фазы вводят газовым шприцем в насадочную или капиллярную колонку хроматографа. Таким методом определяют, например, летучие растворители в почве. Еще один вариант парофазного анализа реализуется, когда не дожидаясь фазового равновесия продувают сосуд с образцом инертным газом. Выдуваемые компоненты собираются на адсорбенте (например, на тенаксе) и вводят в газовый хроматограф после термодесорбции. Пробы растений отбирают на тех же участках, что и пробы почвы. Объединенная проба имеет массу 0, 5-1 кг. Для ее получения отбирают 8-10 точечных проб с пробных площадок, закладываемых по маршруту отбора проб почвы. Площадки для разных сельскохозяйственных культур могут быть размером 1 х 1 м (культура сплошного сева), 1 х 2 м (пропашные культуры). Наземную часть растений срезают острым ножом или ножницами на высоте 3-5 см от поверхности почвы, укладывают в полиэтиленовую пленку или бумагу, вкладывают этикетку. При отборе проб корнеплодов, клубнеплодов и картофеля их следует укладывать для транспортировки отдельно от ботвы. Образцы растений часто измельчают в молотковой дробилке или шаровой мельнице. Следует учитывать возможное загрязнение проб почвой.
ПОКАЗАТЕЛИ ЗАГРЯЗНЕНИЯ ВОД Ранее было отмечено, что стратегия покомпонентного анализа токсикантов применительно к объектам окружающей среды не подходит. Компонентов слишком много, разные значения ПДК, требуются разные методы. Поэтому стремятся найти общие характеристики (показатели) качества воды, которые должны легко определяться в любых лабораториях, должны давать приблизительную оценку качества воды, должны надежно сигнализировать о содержании вредных веществ в воде. Наличие таких показателей не устраняет необходимость определять в воде индивидуальные соединений. Но при постоянном контроле за общими показателями более сложные индивидуальные соединения в лаборатории можно будет определять значительно реже. К общим (интегральным) химическим показателям относятся: мутность, цветность, вкус и запах, удельная электропроводность, общее содержание азота и фосфора, общий органический углерод (ООУ), растворимый органический углерод (РОУ), ХПК, БПК, рН, жесткость. Такое деление на интегральные и индивидуальные показатели условно. Естественно, что изменение химического состава природных вод под действием антропогенных факторов изучают в основном на основании индивидуальных компонентов, а не на основании изменения обобщенных интегральных показателей. Особо следует подчеркнуть, что обобщенный показатель качества воды должен обладать свойством интерпретируемости применительно к оценке качества воды, т.е. иметь вполне однозначно трактуемое смысловое содержание. Многие из применяющихся сейчас в качестве обобщенных показателей условиям однозначной интерпретируемости не отвечают. Так, для правильной оценки качества воды очень важен показатель окисляемости, смысл которого – показать степень возможного изъятия растворенного кислорода из воды водоема содержащимися в ней соединениями. Потреблять кислород могут любые восстановители, а не только органические соединения. Поэтому определение понятия окисляемости (ХПК) как количества кислорода (или окислителя в пересчете на кислород), необходимого для полного окисления содержащихся в пробе органических веществ, и аналогичное определение для БПК неверно передает смысл показателя и может привести к ошибочной интерпретации результатов анализа. Кроме того, применяющиеся сейчас показатели окисляемости дают несопоставимые результаты. Это же можно сказать и о разных методах определения показателя ХПК (табл. 4.5).
Таблица 3. Окисляемость некоторых веществ (в % от теоретической) в зависимости от метода определения
Следовательно, при характеристике качества воды необходимо четко оговаривать, о какой окисляемости идет речь. Показатель БПК (биохимическое потребление кислорода) вообще характеризует возможность существования в воде и аэробного развития колонии бактерий активного ила и способность присутствующих в воде веществ окисляться этой колонией. Как способ оценки окисляемости компонентов сточной воды метод БПК был введен около 160 лет тому назад, когда сточные воды были почти исключительно бытовыми, не содержали трудноокисляемых и сильнотоксичных веществ. Сейчас же определение показателя БПК в классическом варианте лишено смысла из-за невозможности однозначной интерпретации результатов. Необходима замена БПК на два самостоятельных теста: биотест на выживание бактериальной флоры и определение окисляемости в системе с окислительно-восстановительным потенциалом, близким к потенциалу биологической системы. Естественным и, видимо, единственным способом оценки возможного биологического влияния компонентов вод является биотестирование, поскольку химический анализ не позволяет оценить кумулятивные, синергетические и антагонистические эффекты загрязнителей.
Общий азот Общий азот складывается из суммы аммонийного, нитратного и нитритного азота. Азотом органическим при этом, как правило, пренебрегают. Классический метод Дюма определения суммы органического и неорганического азота можно представить в виде следующих стадий: 1) сжигание в токе кислорода с катализатором CuO с образованием оксидов азота; 2) восстановление оксидов на медном катализаторе до N2; 3) улавливание газообразного азота на молекулярном сите; 4) температурно-программированный нагрев концентрата; 5) газохроматографическое определение азота с детектором по теплопроводности.
Удельная электропроводность Электропроводность обусловлена наличием в воде шести основных ионов. Ионы примесных компонентов содержатся в незначительных количествах и ими обычно пренебрегают. Т.е. электропроводность в какой-то мере является характеристикой общей минерализации пробы. Электропроводность определяют кондуктометрическим методом. Ее измеряют методом переменного тока с использованием платиновых электродов с большой поверхностью (платиновая чернь), чтобы уменьшить поляризацию. Сопротивление раствора прямо пропорционально расстоянию между электродами l и обратно пропорционально площади поверхности электродов в растворе S:
электролитической ячейки. Константу электролитической ячейки определяют по 0, 01 М раствору КCl (χ = 0, 001412 См/см) или по 0, 1 М раствору КCl (χ = 0, 01289 См/см). Анализируемую воду наливают в ячейку и термостатируют при (25, 0±0, 1)º С. рН Согласно международному стандарту ИСО 10523 измерение рН проводят электрохимическим методом, основанным на измерении э.д.с. электрохимической ячейки, состоящей из пробы воды, стеклянного электрода и электрода сравнения (чаще всего – хлорсеребряного). Уравнение Нернста для стеклянного электрода:
Величина E0 зависит от свойств стекла. Поэтому электрод надо калибровать по буферным растворам. Результат измерения рН зависит от температуры. В современных приборах иономера-рН-метрах предусматривается автоматическая термокомпенсация. Электрод работает при рН = 1-10. При рН менее 1 наблюдается кислотная погрешность, которая зависит от сорта стекла, а при рН более 10 – щелочная погрешность (электрод становится чувствительным к ионам щелочных металлов). Существуют сорта стекла на основе Li2O-BaO-La2O3-SiO2, для которых диапазон измерения шире как в кислой, так и в щелочной области. Отбор проб для определения рН следует проводить в стеклянные бутылки из боросиликатного стекла с плоским дном и объемом не менее 500 мл. Пластиковые бутыли не рекомендуются из-за их газопроницаемости. Измерение рН пробы воды следует проводить как можно быстрее, т.к. рН быстро меняется вследствие протекания различных химических, физических и биологических процессов в пробе. Если невозможно определить рН воды в месте взятия пробы, ее можно хранить не более 6 часов в заполненной доверху бутыли. При контроле рН суспензии следует осадить осадок, и измерять рН необходимо у чистой фракции. При контроле сточных вод существует высокий риск загрязнения измерительных электродов и мембран маслом, жиром, нефтепродуктами. Для электродов сравнения их загрязнение может быть предотвращено поддержанием избыточного давления раствора хлористого калия. Жесткость Международный стандарт ИСО 6059 устанавливает титриметрический комплексонометрический метод определения суммарной концентрации ионов кальция и магния (жесткость волы) в грунтовых и поверхностных водах, а также в питьевой воде. Метод не применим для минерализованных вод и морской воды. Наименьшая определяемая концентрация составляет 0, 05 ммоль/л. Метод заключается в комплексонометрическом титровании ионов кальция и магния (Ме2+) трилоном Б (HY3–) при рН 10. В качестве индикатора используют эриохром черный Т (HInd2–). Механизм действия индикатора иллюстрируют следующие реакции:
Пробы не требуют предварительной обработки, за исключением тех, которые необходимо профильтровать из-за большого количества взвешенных частиц. Фильтруют пробы через фильтр размером пор 0, 45 мкм сразу же после отбора. При фильтрации существует риск удаления части кальция и магния. Если исследуемые пробы были подкислены для консервации, их нейтрализуют 2 М раствором гидроксида натрия. Ионы металлов – алюминия, бария, свинца, железа, кобальта, меди, марганца, олова и цинка – мешают определению, так как они титруются вместе с ионами кальция и магния и влияют на установление точки эквивалентности. Ионы ортофосфата и карбоната могут осаждать кальций при рН титрования. Анализу могут мешать некоторые органические вещества. Если мешающие влияния невозможно устранить, то анализ проводят пламенным атомно-абсорбционным методом по ИСО 7980. Этот метод применяется для анализа природных и питьевых вод и может быть использован для вод, содержащих Са(II) до 50 мг/л и Mg(II) до 5 мг/л. При десятикратном разбавлении анализируемых проб оптимальная область определения 3-50 мг/л для кальция и 0, 9-5 мг/л для магния. Для устранения анионного матричного влияния и ионизационных эффектов к анализируемой воде добавляют спектрохимические буферы – хлорид лантана LaCl3 (в случае применения ацетилен-воздушного пламени) или хлорид цезия CsCl (для пламени закись азота – ацетилен). Пробы отбирают в чистые полиэтиленовые или полипропиленовые бутыли. Сразу после отбора пробы должны быть подкислены 8 мл концентрированной HCl на каждый 1 л пробы, что препятствует осаждению карбоната кальция. Анализ должен быть проведен сразу после отбора пробы. Согласно ГОСТ 6055-86 жесткостью воды называется свойство воды, обусловленное содержанием в ней ионов кальция (½ Са2+) и магния (½ Mg2+); единицей жесткости воды является моль/м3. В практике чаще используют единицы ммоль/л. В России, начиная с 1952 года, жесткость воды для технических и гигиенических нужд выражается в мг-экв/л, в других странах принято обозначать жесткость в условных градусах. Соотношение между различными единицами жесткости воды, принятыми в разных странах, иллюстрируют данные табл. 4.6.
Как пользоваться этой таблицей? Допустим, что из лаборатории вы получили результаты анализа аквариумной воды: " Общая жесткость С(½ Са2+, ½ Mg2+)" = 3, 25 ммоль/л. Вам надо перевести эту величину в немецкие градусы. В ячейке, соответствующей пересечению строки ммоль/л и столбца немецких градусов находим коэффициент, он же множитель, равный 2, 804. Теперь надо умножить 3, 25 на 2, 804. Произведение этих чисел и будет жесткостью в немецких градусах (dHG). В нашем случае жесткость воды в dGH=9, 11. То есть, сравнительно с ммоль/л, немецкие градусы – более мелкие единицы измерения. Если же вы счастливый обладатель американского теста, и он выдал результат, к примеру, 14 американских градусов (usH), а вам нужны все те же немецкие, то ответ в dGH будет: 14× 0, 056=0, 780. Но это только в том случае, если мы считаем что американский градус равен 1 мг CaCO3 в 1 л воды (так пишут во всей русскоязычной литературе), сами же американцы считают, что их градус жесткости в 17, 12 раз больше, соответственно, и результат измерения в dGH будет равен 13.35. То есть эти американские градусы довольно близки к немецким. Пользование разными единицами измерения жесткости без их пересчета может привести к существенному искажению данных. Так, 14 американских градусов – это всего лишь 0, 78 немецких. Поэтому, читая сообщения американского коллеги-рыбовода, о том, что его рыбки отнерестились при 14° градусах жесткости, не надо думать, что им подходит для нереста жесткая вода, эта вода на самом деле очень мягкая. Биотестирование Химический анализ не позволяет оценить кумулятивные, синергетические и антагонистические эффекты загрязнителей. Эти эффекты исследуют с применением методов биотестирования. Биотестирование вошло в практику в начале 20 в. («рыбная проба») для токсикологических исследований вод. Первые биотесты на дафниях и циклопах были выполнены в 1918 г. Сейчас для биотестирования используются дафнии магна, гидробионты с разными трофическими связями, простейшие, ракообразные, черви, рыбы. Биологическими показателями оценки качества воды служат: выживаемость, размножение, выживаемость нарождающейся молоди, дыхательный и сердечный ритмы, потребление кислорода, выделение углекислого газа и аммиака как конечных продуктов обмена, дыхательный коэффициент, темпы роста и питания, кормовой коэффициент и т.д. В настоящее время биотесты введены в стандарты на качество воды во многих странах. Биотесты с рыбами и ракообразными включены в международные стандарты по оценке качества воды, в частности ИСО 7346, ИСО 6341 и ИСО 10706. Существуют статические и динамические тест-системы, которые позволяют определять острое и хроническое токсикологическое влияние. Из полученных в результате биотестирования данных рассчитывают ЛД50 или ЛК50. В Германии для тестов на приспосабливаемость в случае регулярных выпусков сточных вод используют форель («zebra fish»). Пять рыб помещают в пятилитровый аквариум, рН воды строго 7, 0±0, 2. Температура (20±1)º С, концентрация растворенного кислорода 4 мг/л. Запускают в аквариум сточную воду и выдерживают 48 часов. К концу этого времени определяют, сколько рыб выжило во время теста. Обязательно ставят контрольный опыт – рыб выдерживают в воде, которая используется для разбавления сточной воды. Результат теста выдается в виде фактора разбавления GF, который в Германии означает токсичность и показывает, во сколько раз сточную воду надо разбавить, чтобы приготовить безвредную тестовую среду. Обычно тест в острой форме длится 24 часа, а тест жизненного цикла – 3 недели. Определение только острой токсичности не может дать полной картины. Необходим учет отдаленных эффектов при попадании токсикантов в организм из воды. Биотестирование позволяет изучать влияние только форм веществ, существующих в воде в момент анализа, и не учитывает результаты в случае трансформации компонентов. ОПРЕДЕЛЕНИЕ МЕТАЛЛОВ ИСО 11885 устанавливает метод определения растворенных и нерастворенных элементов, а также их общего количества в питьевой, природной и сточной водах методом атомно-эмиссионной спектроскопии с индуктивно-связанной плазмой. Данным методом можно определять 33 элемента: Ag, Al, As, B, Ba, Be, Bi, Ca, Cd, Co, Cr, Cu, Fe, Li, Mg, Mn, Mo, Na, Ni, P, Pb, S, Sb, Se, Si, Sn, Sr, Ti, V, W, Zn, Zr. Атомно-эмиссионная спектроскопия с индуктивно-связанной плазмой рекомендована при многоэлементном анализе питьевой воды в России (ГОСТ Р 51309-99). Метод основан на измерении интенсивности излучения атомов определяемых элементов, возникающего при распылении анализируемой пробы в аргоновую плазму, индуктивно возбуждаемую радиочастотным электромагнитным полем. Напомним, что качественный анализ основан на характеристичности излучения свободных атомов и ионов в газообразном состоянии Δ E = hν. Количественный анализ основан на зависимости интенсивности излучения от концентрации в соответствии с формулой Ломакина I = a·Cb. Определение общего количества элементов проводят следующим образом. Пробу сразу после отбора (или во время отбора) подкисляют азотной кислотой до рН = 2 или менее. Пробу не фильтруют. Анализ проводят как можно быстрее. Если образовался осадок, его растворяют добавлением кислоты при нагревании. Холостую пробу обрабатывают аналогично. При определении растворенных элементов пробу воды фильтруют через мембранный фильтр с порами 0, 45 мкм. Первые 50-100 мл фильтрата используют для ополаскивания колбы. Фильтрат подкисляют 1 мл концентрированной азотной кислоты до достижения рН< 2. При определении элементов, находящихся во взвешенном состоянии измеренный объем незаконсервированной пробы фильтруют через мембранный фильтр сразу после отбора. Фильтр с частицами переносят в контейнер для хранения и транспортировки. Далее мембранный фильтр помещают в стеклянный стакан, добавляют 4 мл азотной кислоты. Стакан закрывают часовым стеклом и осторожно нагревают до полного растворения осадка на фильтре. Когда кислота почти выпарится, стакан охлаждают. Затем добавляют 3 мл азотной кислоты и снова упаривают пробу. После охлаждения в пробу добавляют 10 мл соляной кислоты и 15 мл воды и аккуратно растворяют все осадки при осторожном нагреве. Пробу охлаждают, часовое стекло и стенки стакана промывают водой. Фильтруют для удаления нерастворенных частиц и разбавляют в зависимости от концентрации определяемых элементов. Определение выполняют по методу градуировочного графика. При определении общего содержания металлов в природных и сточных водах, содержащих значительное количество комплексообразующих веществ, органических соединений, в ряде методик предусматривается предварительное вскрытие таких проб. Описано несколько способов предварительной пробоподготовки. Первое – это мокрое озоление, которое включает 20-тиминутное кипячение пробы с HCl и (NH4)2S2O8 или обработку пробы при кипячении смесью концентрированных серной и азотной кислот с последующим выпариванием до паров серной кислоты. Второй способ предусматривает фотохимическую деструкцию комплексных соединений ионов металлов с органическими соединениями. К пробе воды добавляют серную кислоту и пробу помещают под УФ лампу на 50-60 минут. Этой обработкой достигается разрушение, например, достаточно прочных комплексов макро- и микроэлементов природных вод с природными органическими лигандами – фульвокислотами (табл. 4.8).
Таблица 4.8. Условные константы устойчивости комплексов макро- и микроэлементов вод с фульвокислотами
Значительная интенсификация пробоподготовки природных и сточных вод достигается при деструкции органических соединений с использованием ультразвука (18-44 кГц, 1-2 минуты). Пламенная эмиссионная спектроскопия используется для определения щелочных и щелочноземельных элементов. Так, ИСО 9964-3 устанавливает пламенно-фотометрический метод определения натрия и калия в неочищенной и питьевой воде. Не менее широко в национальных и международных стандартах представлена атомно-абсорбционная спектроскопия. Например, ИСО 8288 устанавливает три метода определения Cd, Ni, Cu, Pb и Zn в воде пламенной атомно-абсорбционной спектрометрией: метод А – прямое определение; метод В – определение после экстракционного извлечения хелатов анализируемых металлов с 1-пирролидиндитиокарбаматом аммония метилизобутилкетоном; метод С – определение после экстракционного извлечения хелатов анализируемых металлов при рН 2-4 с гексаметиленаммонием-гексаметилендитиокарбаматом смесью растворителей диизопропилкетон-ксилол. Метод А применяют, когда концентрации анализируемых элементов сравнительно велики и нет мешающих влияний. Когда пробы имеют сложную или неизвестную природу или содножат высокие концентрации растворенных минеральных веществ (рассолы или соленые воды), применяют методы В, С. Концентрации элементов, определяемых разными методами, представлены в табл. 4.9.
Таблица 4.9. Рекомендуемые диапазоны концентраций
Градуировочные растворы также проводят через стадию экстракции. Следует особо подчеркнуть, что экстракты неустойчивы и могут храниться только несколько часов. Экстракт кадмия следует анализировать немедленно. Государственный стандарт Российской Федерации ГОСТ Р 51309-99 предусматривает электротермическое атомно-абсорбционное определение в питьевых водах и водах источников водоснабжения Al, Ba, Be, V, Bi, Fe, Cd, Co, Mn, Cu, Mo, As, Ni, Sn, Pb, Se, Ag, Sb, Ti, Cr, Zn. Метод основан на измерении поглощения излучения резонансной длины волны атомным паром определяемого элемента, образующимся в результате электротермической атомизации анализируемой пробы в графитовой печи спектрометра. Метод позволяет определять массовые концентрации (в мг/л):
Пробу воды отбирают в посуду объемом 0, 2-0, 5 л, изготовленную из полимерных материалов. Если измерение проводят более чем через 5 часов после отбора, пробы консервируют, добавляя на 200 мл воды 3 мл концентрированной азотной кислоты. Если в подкисленной воде находятся заметные глазом взвешенные частицы, то перед проведением измерений ее фильтруют. Для устранения матричных влияний используется химический модификатор – смесь Pd(NO3)2-Mg(NO3)2. Для проверки наличия мешающего влияния матрицы из раствора подготовленной пробы отбирают аликвотную часть и разбавляют ее в 5-10 раз 0, 3 М раствором азотной кислоты. Измеряют величину аналитического сигнала определяемых элементов в исходной и разбавленной пробе. Мешающие влияния считаются незначимыми, если выполняется условие:
Для устранения неселективного поглощения используют различные способы коррекции: с использованием непрерывного спектра дейтериевой лампы (дейтериевый корректор фона), на основе эффекта Зеемана и другие. Прежде чем изложить сущность этих способов, необходимо вспомнить устройство оптической схемы атомно-абсорбционного спектрофотометра. Все оптические схемы делят на однолучевые и двухлучевые. В двухлучевых приборах свет от источника первичного излучения делится с помощью зеркального модулятора на два луча, один из которых проходит через атомизатор, а второй – минует его (рис. 4.5).
Рис. 4.5 – Однолучевая (А) и двухлучевая (В) оптические схемы приборов: 1 – источник резонансного излучения; 2 – атомизатор; 3 – монохроматор (дисперсионная система); 4 – модулятор; 5 – полупрозрачное зеркало Перед входом в монохроматор оба луча сводятся с помощью полупрозрачного зеркала. Двухлучевая схема позволяет устранить неконтролируемые изменения (дрейф) в интенсивности источника первичного излучения. Однако она не компенсирует дрейф чувствительности самого атомизатора. После монохроматора дифрагированное излучение попадает на детектор, в качестве которого в атомно-абсорбционной спектроскопии используется фотоэлектроумножитель (ФЭУ). В пламенной атомной абсорбции постоянный сигнал интегрируется за период времени несколько секунд. В электротермической атомной абсорбции сигнал имеет вид пика, измерение которого выполняют как по высоте, так и по площади. При работе со сплошным источником через пламя или графитовую печь поочередно пропускают резонансное излучение и излучение сплошного спектра. В первом случае измеряется суммарное (резонансное и неселективное) поглощение, а во втором – неселективное, и в качестве результата выдается их разность. В области длин волн 190-350 нм в качестве источника сплошного спектра используется дейтериевая лампа, в области 350-770 нм – галогенная лампа. При использовании корректора фона необходима точная юстировка обоих световых пучков, обеспечивающая их прохождение через одну и ту же область атомизатора. Существует предельная величина неселективной абсорбции, выше которой компенсация с помощью дейтериевого корректора невозможна. Эта величина разная для различных типов приборов, например, для прибора «Сатурн – 3П1» она порядка 30% шкалы. При работе с корректором фона возрастает шум, ухудшаются воспроизводимость и предел обнаружения. При компенсации неселективной абсорбции с тонкой структурой спектра возможны систематические погрешности. Автоматический корректор компенсирует усредненное значение неселективной абсорбции в данном спектральном интервале, которое может не совпадать с ее значением на длине волны резонансной линии. Так, при определении золота в хлориде индия автоматический дейтериевый корректор «перекомпенсирует» неселективную абсорбцию. Правильность работы корректора можно проверить, производя измерения со щелями различной ширины: значения атомной и неселективной абсорбции при этом не должны меняться. Ряд трудностей, возникающих при работе с дейтериевым корректором, может быть успешно преодолен при использовании для компенсации неселективных помех эффекта Зеемана (рис. 4.6).
Рис. 4.6 – Схема учета неселективного поглощения на основе эффекта Зеемана: 1 – лампа с полым катодом; 2 – вращающийся поляризатор; 3 – атомизатор между полюсами магнита (5-15 кГс); 4 – монохроматор; 5 – фотоумножитель; 6 – усилитель; 7 – регистрирующее устройство; 8 – контур p-компоненты линии поглощения; 8′, 8″ – контуры ±s-компонент линии поглощения; 9 – неселективное поглощение; 10 – контур эмиссионной линии.
В постоянном магнитном поле абсорбционная линия расщепляется на три компоненты: p (DМ = 0) и ±s(DМ = ±1), причем p-компонента не смещается относительно центра линии, но поляризована в направлении, параллельном направлению магнитного поля, а ±s- компоненты смещены относительно центра линии и поляризованы перпендикулярно магнитному полю. Соответственно, когда с помощью вращающегося поляризатора на атомизатор от лампы с полым катодом поступает излучение, поляризованное параллельно магнитному полю, будет регистрироваться суммарное поглощение p- компоненты и фона, а когда поступает излучение, поляризованное перпендикулярно магнитному полю, в атомизаторе фиксируется только неселективное поглощение, причем строго на той же длине волны, что и сигнал атомной абсорбции. Разность этих двух измерений дает значение полезного сигнала. Зеемановская коррекция фона позволяет компенсировать неселективное поглощение до уровня 2, 0 единиц абсорбционности. Кроме того, она обеспечивает коррекцию как непрерывного фонового поглощения, так и линейчатого. Преимуществом этого способа коррекции является то, что обеспечивается условие равенства интенсивностей и юстировки 2-х лучей, возможна компенсация неселективного поглощения во всем диапазоне длин волн, компенсируются большие величины неселективного поглощения, в том числе в спектрах поглощения с тонкой структурой. Недостатком использования эффекта Зеемана для коррекции фонового поглощения является небольшое ухудшение линейности градуировочных кривых и незначительная потеря чувствительности для некоторых элементов. Это, однако, компенсируется точностью фоновой коррекции и значительным улучшением соотношения сигнал/шум по сравнению с коррекцией с помощью источника непрерывного спектра. Результатом этого является улучшение воспроизводимости, правильности и пределов обнаружения для реальных образцов. Корректор Смита-Хифти связан с так называемым «эффектом самообращения» резонансной линии, который выражается в том, что при достаточно большом парциальном давлении атомов исследуемого элемента в колбе лампы испущенное возбужденными атомами излучение поглощается невозбужденными атомами, причем в большей степени поглощается центральная часть спектральной линии излучения. Поэтому форма спектральной линии резонансного излучения, выходящего из лампы в условиях самообращения, имеет провал (минимум) на центральной частоте резонансного перехода и максимумы слева и справа от этой частоты. Ширина и глубина центрального минимума зависят от величины тока питания лампы. При этом используют импульсное питание источника резонансного излучения токами различной величины. При достаточно больших токах можно достичь того, что в некоторой окрестности центральной частоты интенсивность выходящего излучения будет равна нулю. Это является необходимым условием использования эффекта самообращения для учета неселективного поглощения. Достигается это следующим путем. На лампу с полым катодом попеременно подают то слабый (обычно от нескольких мА до 30 мА), то сильный ток (до 150-300 мА). При слабом токе форма спектральной линии выходящего из лампы резонансного излучения имеет обычный вид с максимумом на центральной частоте атомного перехода. Поэтому фотоприемник зарегистрирует суммарный сигнал абсорбции, обусловленный резонансным поглощением определяемого элемента и неселективным фоновым поглощением. При сильном токе в лампе в атомизаторе абсорбируется излучение, в спектре которого из-за эффекта самообращения интенсивность равна нулю как раз в некоторой окрестности центральной частоты атомного перехода. Если ширина этой частоты спектра больше спектральной ширины линии резонансного излучения, возникающего при слабом токе, то фотоприемник в этом случае зарегистрирует сигнал абсорбции, соответствующий только неселективному фоновому поглощению. Измерительная система прибора выдает разность сигналов, соответствующих слабому и сильному току, т.е. сигнал, связанный с абсорбцией только атомами определяемого элемента. Этот способ коррекции фона имеет те же плюсы, что и эффект Зеемана, однако здесь эффективность коррекции зависит от качества источника излучения. Кроме того, при такой работе ламп с полым катодом значительно сокращается время их жизни, особенно для легколетучих элементов, таких как свинец, кадмий, цинк и др. Метод атомно-абсорбционной спектроскопии рекомендован для определения в водах калия (ИСО 9964-2), натрия (ИСО 9964-1), мышьяка (ИСО 11969), ртути (ИСО 5666), селена (ИСО 9965), хрома (ИСО 9174). Причем для мышьяка и селена атомно-абсорбционные спектрометры должны быть оснащены гидридным генератором. Сущность метода заключается в следующем. Определяемый элемент переводится в газообразный гидрид действием сильных восстановителей в кислой среде (чаще всего для этих целей используют тетрагидроборат натрия NaBH4). Газообразные гидриды выдуваются инертным газом из реактора, смешиваются в камере с горючим газом (чаще всего с водородом), и полученная гомогенная смесь подается в пламя (рис 1.3). Иногда вместо диффузного воздушно-аргоноводородного пламени с гидридной системой используют пламя динитроксид-ацетилен. Это пламя прозрачно в области от 190 нм, что особенно важно, т.к. резонансные линии основных элементов, определяемых с помощью гидридных систем, лежат в коротковолновой области спектра. Это элементы, образующие легколетучие гидриды, такие, как мышьяк (193, 7 нм), селен (196, 0 нм), сурьма (217, 6 нм), висмут (223, 1 нм), теллур (214, 3 нм), олово (224, 6 нм).
Рис. 4.7. – Схема гидридной системы Основной способ определения ртути в водах – это атомно-абсорбционная спектроскопия методом «холодного пара». Международный стандарт ИСО 5666 устанавливает методы определения общей ртути в воде и состоит из трех частей. Эти части отличаются друг от друга способами подготовки проб для устранения мешающего влияния органических веществ в зависимости от их концентрации в различных типах вод. Часть 1 устанавливает метод анализа путем минерализации перманганатом-персульфатом калия. Метод применим к природным, промышленным, сточным водам и водам, предназначенным для хозяйственно-бытовых нужд. Часть 2 устанавливает метод анализа путем минерализации ультрафиолетовым облучением. Он применим к питьевым водам и водам, предназначенным для приготовления напитков и пищевых продуктов. Часть 3 устанавливает метод анализа путем минерализации бромом. Метод применим к пресным, соленым и питьевым водам, а также к другим типам вод, содержащим небольшое количество органических веществ. Сущность метода заключается в минерализации (химическом озолении) анализируемой пробы с целью удаления всех органических соединений. Избыток окислителя устраняют обработкой пробы солянокислым гидроксиламином и восстанавливают ртуть(II) до металлического состояния обработкой хлоридом олова(II). Отгонку ртути проводят током газа при комнатной температуре и регистрируют атомное поглощение парами ртути резонансного излучения высокочастотной безэлектродной лаипы на ртуть при 253, 7 нм. Наиболее часто для определения ртути используется установка, схему которой можно представить следующим образом (рис. 4.8).
Рис. 4.8. Установка для определения ртути методом холодных паров с замкнутой системой коммуникаций: 1 – реактор; 2 – насос; 3 – фильтр; 4 – кювета; 5 – поглотитель; 6 – краны
Анализируемую пробу предварительно переводят в раствор, в котором ртуть находится в двухвалентном состоянии. Раствор помещают в колбу-реактор (1), куда затем приливают раствор хлорида олова(II), восстанавливающего ртуть до металла: Hg(II) + Sn(II) = Hgo + Sn(IV) Затем с помощью циркуляционного насоса (2) прокачивают находящуюся в системе газовую смесь, содержащую выделившиеся пары ртути, через кювету с кварцевыми окнами (4), устанавливаемую на оптической оси конденсорной системы спектрофотометра вместо пламени или электротермического атомизатора. Для очистки потока газа от частиц аэрозолей служит фильтр (3). Так как система коммуникаций замкнутая, то в ней спустя несколько десятков секунд после начала реакции устанавливается равновесная концентрация паров ртути; при этом атомное поглощение ее аналитической линии (253, 7 нм) достигает максимума (рис. 4.9).
Рис 4.9. Зависимость аналитического сигнала ртути от времени при работе установки методом «холодного пара» с замкнутой системой коммуникаций
Зависимость атомно-абсорбционного сигнала от времени имеет форму кривой с насыщением, причем предел насыщения пропорционален содержанию ртути в растворе пробы. После окончания измерения открывают краны (6) и ртутные пары улавливаются поглотителем (5). Для получения градуировочной характеристики используют растворы с известным содержанием ртути (метод градуировочного графика). Чаще в приставках к атомно-абсорбционному спектрофотометру для определения ртути методом «холодного пара» используют системы с незамкнутой системой коммуникаций. Пример такой установки приведен в ИСО 5666 (рис. 4.10).
Рис. 4.10. Установка для определения ртути (по ИСО 5666): 1 – сжатый воздух или инертный газ; 2 – ротаметр; 3 – дегазационная колба; 4 – аэрационная трубка; 5 – самописец; 6 – спектрофотометр; 7 – кварцевая измерительная камера; 8 – нагреватель; 9 – ртутная лампа; 10 – абсорбер ртути
Система коммуникаций не замкнутая. В процессе определения проходящий через систему воздух с парами ртути поступает в аналитическую кювету. Аналитический сигнал возрастает по мере поступления атомов в ячейку, проходит через
Рис. 4.11. Форма аналитического сигнала при определении ртути методом холодного пара в установках с незамкнутой системой коммуникаций
Варьируя различные восстановители и способы обработки проб, можно дифференцированно определять неорганические, фенильные и алкильные формы ртути при их совместном присутствии в природных водах. Схема анализа представлена на рис. 4.12. После отбора пробы воды и общепринятой мембранной фильтрации для разделения растворенных и взвешенных форм ртути допускается консервирование воды 0, 2 М HNO3 на срок, не более 7 дней. Для определения ртути, связанной в комплексы, lg Куст которых £ 15 (С1), используют восстановление SnCl2 в солянокислой среде. Определение суммы неорганических и органических форм ртути, включая ее гидроксокомплексы с фульво- и гуминовыми кислотами (С2), проводят после предварительной обработки смесью NaCl – HNO3 с помощью SnCl2 в сернокислой среде. С помощью гидразинборана определяют сумму неорганических и арильных соединений ртути (С3). Разница между С3 и С2 соответствует содержанию арильной ртути. После разрушения пробы воды окислительной бромид-броматной смесью или смесью окислителей (HNO3 + H2SO4 + Na2S2O8 + KMnO4) со SnCl2 или гидразинбораном определяют общее содержание ртути (С4). Разница между С4 и С3 соответствует содержанию алкильных форм ртути.
Рис.4.12. Анализ природных вод при определении различных форм ртути Еще одним методом, используемым для определения валового содержания свободных и связанных ионов металлов, является метод инверсионной вольтамперометрии. Например, этот метод включен в нормативные документы: – МВВ 081/12-0290-06. Методика выполнения измерений содержания кобальта, никеля, железа, хрома, серебра, таллия в питьевой воде методом инверсионной вольтамперометрии; – МВВ 081/12-0288-06. Методика выполнения измерений содержания марганца в природной, питьевой и очищенной сточной воде методом инверсионной вольтамперометрии; – МВВ 081/12-4631-00. Методика выполнения измерений содержания кадмия, свинца, меди в природных и очищенных сточных водах методом инверсионной вольтамперометрии; – МВВ 081/12-0139-04. Методика выполнения измерений содержания цинка в природных и очищенных сточных водах методом инверсионной вольтамперометрии; – МВВ 081/12-0094-03. Методика выполнения измерений содержания мышьяка в природной, питьевой и очищенной сточной воде методом инверсионной вольтамперометрии; – МВВ 081/12-0095-03. Методика выполнения измерений содержания ртути в природной, питьевой и очищенной сточной воде методом инверсионной вольтамперометрии; – МВВ 081/12-0200-05. Методика выполнения измерений содержания селена в воде питьевой и природной методом инверсионной вольтамперометрии; Напомним, что вольтамперометрическими называют методы анализа, основанные на регистрации и изучении зависимости тока, протекающего через электролитическую ячейку, от внешнего наложенного напряжения. Типичная зависимость силы тока от приложенного к электролитической ячейке напряжения приведена на рис. 4.13.
Рис. 4.13. Полярограмма: 1 – остаточный ток; 2 – диффузионный ток Если в растворе нет веществ, способных восстанавливаться под действием электрического тока, то сила тока в цепи будет небольшой. Этот ток называют остаточный. Если же в растворе есть вещества, способные восстанавливаться, то при достижении определѐ нного потенциала ионы начнут восстанавливаться на ртутном катоде, нередко с образованием амальгамы: Mn+ + ne + Hg = M(Hg) При этом сила тока в цепи возрастѐ т (полярографическая волна). С этого момента рост потенциала электрода как бы отстает от роста налагаемого внешнего напряжения – электрод деполяризуется. Вещество, участвующее в электрохимической реакции и вызывающее деполяризацию электрода, называют деполяризатором. Ток растѐ т до восстановления всех ионов, находящихся вблизи поверхности электрода. При этом ток практически не зависит от потенциала электрода. Новые порции ионов к поверхности капли из раствора доставляются за счет диффузии. Ток, соответствующий этому потенциалу, называют диффузионным током Id. Восстанавливающиеся ионы могут быть доставлены к электроду также за счет миграции и конвекции. Параметры полярографической волны дают возможность провести качественный и количественный анализ. Потенциал, отвечающий току I=½ Id, называется потенциал полуволны Е½ . Его числовое значение показывает, насколько легко восстанавливается на электроде данное вещество. Это качественная характеристика вещества; потенциал полуволны непосредственно связан со стандартным потенциалом данной окислительно-восстановительной системы. Значение Е ½ зависит от состава фонового электролита в ячейке. Для количественного определения электроактивных веществ используется прямая пропорциональная зависимость между диффузионным током (или высотой волны) и концентрацией деполяризатора. Зависимость диффузионного тока Id от концентрации иона С выражается уравнением Ильковича:
где Id – диффузионный ток; n – число электронов, принимающих участие в электродной реакции; D – коэффициент диффузии определяемого вещества; m – скорость вытекания ртути из капилляра; τ – время образования одной капли; С – концентрация деполяризатора. Существенное увеличение чувствительности дает инверсионная вольтамперометрия. Сущность этого метода состоит в выделении определяемого элемента из очень разбавленного раствора на ртутной капле или на графитовом электроде электролизом с последующим анодным растворением полученной амальгамы. Зависимость силы тока от напряжения при анодном растворении имеет вид характерного пика, глубина которого h пропорциональна концентрации определяемого иона (hmin = k ·C), а потенциал минимума Emin определяется природой иона (рис. 4.14):
Рис. 4.14. Кривая анодного растворения
Предел обнаружения в методике инверсионной вольтамперометрии на 2-3 порядка ниже предела обнаружения в обычных полярографических методиках. Нижняя граница определения методом инверсионной вольтамперометрии, например, Cu, Zn, Pb, Cd составляет 0, 1-1 мкг/л. При определении общего содержания металлов пробу воды вскрывают кислотами или используют ультразвуковую обработку. Свободные ионы металлов определяют непосредственно в пробе без деструкции органических соединений. Содержание ионов металлов, связанных в комплекс, находят как разницу между их общим содержанием и содержанием свободных ионов.
ОПРЕДЕЛЕНИЕ ПОВЕРХНОСТНО-АКТИВНЫХ ВЕЩЕСТВ (ПАВ) Поверхностно-активные вещества – высокомолекулярные соединения, концентрирующиеся на поверхности раздела фаз, вызывая снижение поверхностного (межфазного) натяжения. Анионоактивные ПАВы – сульфаты и сульфонаты: R–SO3–Na(K) R–O–SO3–Me Катионные ПАВы:
R1 – C12-C18; R2, R3, R4 – метильный, этильный или фенильный радикалы; An– - Cl–, Br–, I–. Неионогенные ПАВы: R–X–(CH2–CH2O)mH, где R – алкильный радикал; Х – атомы O, S или группы –COO–, –CONH– ПАВы в водах определяют либо сразу после отбора проб либотконсервируют добавлением 2-4 мл хлороформа на 1 литр и хранят приттемпературе 3-5º С. Стандартными являются несколько методов определения: – титриметрический; – полярографический; – спектрофотометрический. Международный стандарт ИСО 7875 предусматривает экстракционно- фотометрический метод определения анионных ПАВ, которые часто входят в состав синтетических моющих средств. Сущность метода заключается в образовании в щелочной среде ассоциатов анионных ПАВов с метиленовым синим и экстракции этих ассоциатов хлороформом с последующей обработкой хлороформного экстракта кислотой:
Экстракт фотометрируют при 650 нм. Сам метиленовый синий в хлороформе не растворяется. Экстракцию ионного ассоциата проводят при рН = 10. Определению мешают S2–, SO3 2– и другие вещества, которые восстанавливают метиленовый синий, их предварительно окисляют. Определению мешают также катионные ПАВы. Современной модификацией этого метода является использование отражательной спектроскопии. Анионные ПАВы образуют ионные ассоциаты с катионом ферроина:
Далее следует сорбция этого ионного ассоциата, а затем конечное определение методом диффузного отражения. В отражательной спектроскопии используется рассеяние света – распространение излучения от поверхности непрозрачного тела во всех направлениях. Рассеяние света будет равномерным, если частицы рассеивающих центров находятся близко друг к другу, как в случае тонкодисперсных порошков. Возникающее при этом явление называется диффузным отражением. Его мерой служит отношение интенсивностей рассеянного (I) и падающего (I0) излучения:
Спектр диффузного отражения подобен спектру поглощения вещества в растворе. Эти спектры можно непосредственно использовать для идентификации твердых непрозрачных окрашенных образцов – пигментов, порошков, слоев краски, поверхностей металлов. Пламенно-ионизационный детектор (ПИД) Пламенно-ионизационный детектор (ПИД) основан на ионизации органических соединений в пламени водорода. Устройство детектора представлено на рис. 4.18.
Рис. 4.18. Схема пламенно-ионизационного детектора: 1 – ввод газа из колонки; 2 – ввод воздуха; 3 – катод; 4 – собирающий электрод; 5 – вывод в атмосферу Выходящий из колонки газ смешивается с H2 и поступает в форсунку горелки детектора. Образующиеся в пламени ионизованные частицы заполняют межэлектродное пространство, в результате чего сопротивление снижается, ток резко усиливается. Т.е. концентрацию ионов определяют, измеряя проводимость пламени. ПИД реагирует практически на все соединения, кроме H2, инертных газов, О2, N2, оксидов азота, S, C, а также H2О. Этот детектор чувствителен только к соединениям, ионизирующимся в пламени, т.е к соединениям с C-C и C-H связями. Точный механизм ионизации не выяснен. С использованием масс- спектрометрометрии проведено исследование и обнаружено, что механизм ионообразования связан с термодеструкцией и последующей хемоионизацией. В ПИД одним из электродов служит горелка, второй электрод – коллектор – располагается над горелкой. Малые токи (10–9–10–12 А) усиливаются, т.к. шумы самого детектора малы. Из-за высокой чувствительности, большого диапазона линейности ПИД стал наиболее распространенным детектором. Термоионный детектор (ТИД) ТИД селективен к N- и P-содержащим соединениям за счет введения в пламя водорода паров солей щелочных металлов (К, Na, Rb и Cs). Скорость введения паров щелочных металлов должна быть стабилизирована. ТИД чувствителен к стабильности поддержания скорости водорода, воздуха и газа-носителя. Селективность ТИД к N- и P- органическим соединениям по сравнению с ПИД — порядка 102–103. Пламенно-фотометрический детектор (ПФД) ПДФ селективен к S- и P-содержащим соединениям, при сжигании которых в пламени, обогащенном водородом, по сравнению с ПИДом, излучаемый свет от этих элементов направляется на фотоумножитель через специальные фильтры (394 нм для S и 526 нм для Р). Особенности детектора: · чувствительность ПФД к S-и Р-содержащим соединениям тем больше, чем выше содержание этих элементов в соединениях; · сигнал к Р-содержащим соединениям пропорционален концентрации этого вещества в газе-носителе; · сигнал к S-содержащим соединениям пропорционален логарифму потока вещества. Фотоионизационный детектор (ФИД) В ФИДе ионизация анализируемых соединений происходит за счет УФ-излучения в специальной камере с двумя электродами. При фотоионизации молекулы анализируемых соединений диссоциируют на ион и электрон: А + h ν → A+ + е– Образуемые ионы собираются электродами. Ионизируются только те соединения, потенциал которых ниже энергии фотонов. В зависимости от лампы энергия фотонов может быть 9, 5; 10, 2 и 11, 7 эВ. ФИД как и ПИД обладает высокой чувствительностью ко всем органическим соединениям. К ароматическим соединениям ФИД имеет в 10–50 раз большую чувствительность, чем ПИД. В отличие от ПИД фотоионизационный детектор может регистрировать H2S, PH3, NH3, AsH3 и др. Масс-спектрометрический детектор (МСД) В последние годы достигнут прогресс в создании небольших настольных МСД для газовых хроматографов. В настоящее время этот высокочувствительный детектор — самый совершенный прибор для идентификации неизвестных веществ. Имеется библиотека относительных масс для более 250 тысяч соединений. МСД обычно включает вакуумный насос, ионный источник и систему обработки. Для газовых хроматографов используются в основном два вида ионизации: электронный удар и химическая ионизация. В качестве анализатора ионов могут применяться магнитные, квадрупольные и ионные ловушки, анализаторы ионно-циклотронного резонанса, с двойной фокусировкой (магнитные и электростатические), времяпролетные. В качестве детектора, регистрирующего пучки ионов, используются электронный и фотоэлектронный умножитель, коллектор Фарадея, плоская электронная матрица. МСД – ионизационный деструктивный потоковый детектор, универсальный и одновременно селективный, т.к. всегда можно найти массу, типичную только для данного соединения. При исследовании МСД в режиме детектирования отдельных ионов чувствительность его очень высока (в 1000 раз больше, чем в режиме сканирования) около 10–13 г. Международный стандарт ионизации 70 эВ (1, 1 · 10–17 Дж) общепризнан, на многих современных хромато-масс-спектрометрах предусмотрен только такой фиксированный режим ионизации. Создана библиотека масс с этим источником ионизации.
ИНДЕКС ЧЕРНОГО ДЫМА В настоящее время ¾ взвешенных частиц в атмосфере имеют промышленное или бытовое происхождение. Из стационарных источников аэрозолей наибольший вклад в загрязнение воздуха вносят теплоэлектроцентрали, заводы черной и цветной металлургии, мусоросжигательные печи, бытовые отопители и др. В городах основным источником аэрозолей являются автомобили. Качественный метод определения загрязнения воздуха частицами сажи (индекс черного дыма) устанавливает международный стандарт ИСО 9835. Сущность метода заключается в пропускании воздуха через фильтровальную бумагу и измерении ее светопропускания для оценки загрязненности воздуха частицами сажи. Материал фильтра должен обеспечивать практически 100%-ное улавливание частиц размером от 0, 1 до 5 микрон. Устанавливают рефлектометр на 100%-ное отражение (нулевую абсорбцию) на чистой фильтровальной бумаге, а затем чистую бумагу заменяют экспонированным листом из держателя фильтра и измеряют отражение.
ГРАВИМЕТРИЧЕСКИЙ МЕТОД ОПРЕДЕЛЕНИЯ ВЗВЕШЕННЫХ ЧАСТИЦ Метод определения в воздухе разовых и среднесуточных концентраций взвешенных частиц пыли от 0, 06 до 10 мг/м3 устанавливает стандарт СЭВ 3846-82. Сущность метода заключается в определении массы частиц пыли, задержанных фильтром при прохождении через него определенного объема воздуха. Гидрофобный фильтр (например, перхлорвиниловый АФА-В или ФПП-15-1, 5) задерживает частицы диаметром от 1 до 100 мкм. Фильтр сушат до постоянной массы до отбора пробы и после отбора и взвешивают на аналитических весах.
ОПРЕДЕЛЕНИЕ АСБЕСТА Опасность для человека представляют присутствующие в воздухе асбестовые волокна определенного размера. Поэтому стандартным методом определения содержащегося в воздухе асбеста (ИСО 10312) является просвечивающая электронная микроскопия, позволяющая благодаря своему высокому разрешению идентифицировать даже мельчайшие волокна асбеста. Сущность метода заключается в покачивании пробы воздуха определенного объема через капиллярно-пористый фильтр из поликарбонатной или нитратно-целлюлозной смолы с размером пор 0, 4-0, 45 мкм. Затем на поверхность фильтра напыляют в вакууме углерод и полученную реплику помещают на микроскопические сетки с определенным количеством отверстий и исследуют на электронном микроскопе при увеличении 10000х – 20000х. Чувствительность анализа – 0, 1 волокно/литр воздуха.
ОПРЕДЕЛЕНИЕ МЕТАЛЛОВ Тяжелые металлы поступают в воздух в виде паров или в твердом состоянии. В газообразном состоянии в воздухе находятся металлы с высокой плотностью паров, например, ртуть. Более распространенной формой существования металлов в воздухе являются аэрозоли и пыли. В общей эмиссии пыли топливно-энергетическая промышленность дает около 60% пыли, в то время как черная металлургия – около 10%. Пыль содержит такие металлы, как свинец, кадмий, никель, медь, цинк, хром и__др. В городах среди загрязнений воздуха металлами наибольшая доля приходится на свинец, частицы которого присутствуют в выхлопах двигателей автомобилей. Добавки к топливу тетраэтилсвинца (С2Н5)4Pb (ТЭС) улучшают антидетонационные свойства топлива и увеличивают его октановое число; после сгорания топлива в выхлопных газах содержится аэрозоль частичек свинца, состоящий на 80% из частиц диаметром около 0, 9 мкм. В настоящее время продажа этилированного бензина на Украине запрещена. Содержание многих цветных металлов в воздухе рабочей зоны может быть превышено на сварочных участках, где в воздух поступают пары сварочных аэрозолей.
ДРУГИЕ МЕТАЛЛЫ, РТУТЬ Эмиссионный спектральный анализ является одним из основных методов определения следовых количеств тяжелых металлов в объектах окружающей среды. Стандартные методики Госкомсанэпиднадзора России предусматривают использование атомно-эмиссионного метода при определении в воздухе рабочей зоны Sn, Pb, Cu, Cr, Ni, V, Al, Mg, Zn, Sb, Ti, Li, Si, оксидов РЗЭ, а в атмосферном воздухе – Be, Sn, Pb, Cd, Cu, Cr, Ni, Co, Bi, Mo, V, Fe, Al, Ca, Mg. Анализируемый воздух аспирируют через аэрозольные фильтры (обычно АФА-ХА-18 или АФА-ХП-18) с определенной скоростью в течение времени, указанного в методике. Фильтры переносят в фарфоровые тигли, добавляют 10-15 мг графита (коллектор) и сжигают их в муфельной печи при температуре 450-600º С. В ряде методик озоление проб проводят смесью кислот. Пробу охлаждают, переносят на часовое стекло и добавляют графитовый порошок. Полученную смесь тщательно перемешивают, набивают в кратер графитового электрода и анализируют на кварцевом спектрометре типа ИСП-30 с использованием в качестве источника возбуждения спектров дуги переменного тока. Атомно-эмиссионный метод с источником возбуждения в видеэлектрического разряда в последнее время практически полностью вытеснен индуктивно-связанной плазмой (ИСП). ИСП-спектроскопия во многих странах является одним из основных методов определения металлов в воздухе, воде, почве. На ее основе разработаны стандартные методики, утвержденные на государственном уровне, определения в воздухе рабочей зоны промышленных предприятий следовых количеств Ta, Hf, Ag, B и многих других металлов и их соединений. ИСП-плазма образуется в горелке за счет индукционного нагрева аргона током высокой частоты (40-50 МГц) и поджигается автоматически с помощью искры. Горелка изготовлена из кварца и снабжена инжектором из оксида алюминия, устойчивым к воздействию любых кислот, включая плавиковую кислоту и царскую водку. Пробу в виде аэрозоля впрыскивают в центральную зону горелки с помощью распылителя. Спектрометр оснащен полихроматором (например, Эшелле полихроматор со скрещенной дисперсией). Разрешение оптической системы спектрометра очень высокое – 0, 007 нм при длине волны 200 нм. Детектор на основе фотодиодной матрицы (более 6000 ячеек на кремниевой подложке). Конструкция датчиков позволяет одновременно измерять параметры более чем 5000 спектральных линий, включая измерение фона. К стандартным методам определения приоритетных металлов- загрязнителей воздуха (Hg, Cr, Ni, Co, Cd, Cu, As, Pb, Be и др.) относится также атомно-абсорбционная спектроскопия. Низкие пределы обнаружения, высокая селективность и доступность аппаратуры сделали этот метод одним из главных методов определения металлов в воздухе, воде и почве. Более 25 элементов и их соединений определяется в воздухе в соответствии со стандартными атомно-абсорбционными методиками. Определение, как правило, основано на концентрировании металлов и их соединений на ацетилцеллюлозных фильтрах (например, АФА-ХП-18), кислотном разложении фильтров (смеси азотной и серной кислот, азотной кислоты и пероксида водорода, царская водка), растворении остатка в растворе азотной кислоты и определении содержания определяемых металлов в этом растворе атомно-абсорбционным методом. Атомно-абсорбционный метод обеспечивает чрезвычайно низкие пределы обнаружения паров ртути в воздухе – нижний предел обнаружения 0, 0001 мг/м3, диапазон определяемых концентраций 0, 0001- 0, 004 мг/м3. Анализируемый воздух прокачивают через поглотительный раствор (смесь азотной кислоты и дихромата калия) для окисления ртути и переведения ее в форму Hg2+: 3Hg + Cr2O72– + 14H+ = 3Hg2+ + 2Cr3+ + 7H2O Аликвоту полученного раствора вносят в реактор анализатора ртути (известны модели «Юлия», «Ртуть»), добавляют восстановитель (SnCl2) и пары ртути прокачивают через кювету, где и происходит поглощение резонансного излучения атомными парами ртути: Hg2+ + Sn(II) = Hg↑ + Sn(IV) Наряду с атомно-абсорбционным методом описаны многочисленные спектрофотометрические методы определения соединений металлов в воздухе.
ОПРЕДЕЛЕНИЕ O3 Мониторинг содержания озона в воздухе необходим как в приземном слое воздуха, так и в стратосфере. Причем, если вблизи земли в окружающем воздухе содержание озона должно быть как можно меньше, то в стратосфере на высоте примерно 24 км уменьшение содержания озона в так называемом озоновом слое (истощение) относится к трем важнейшим проблемам загрязнения атмосферы наряду с накоплением парниковых газов и формированием кислотных дождей под воздействием выбросов оксидов серы. Международный стандарт ИСО 10313 регламентирует хемилюминесцентный метод определения массовой концентрации озона в окружающем воздухе в диапазоне от 2 мг/м3 (0, 001 ppm) до 10 мг/м3 (5 ppm). Сущность метода заключается в определении озона при хемилюминесценции пробы при ее обработке этиленом. Интенсивность выделяемого при реакции света (λ = 253, 7 нм) пропорциональна концентрации озона в пробе воздуха:
Современные дистанционные методы определения озона в верхних слоях атмосферы основаны на принципах молекулярного абсорбционного анализа: O3 поглощает солнечный свет при 303-315 нм, NO2 – при 437-443 нм, пары H2O и CO2 поглощают в ИК области при 4879-4910 см–1. При спектральном разрешении 0, 7 нм записывается спектр в интервале 303-315 нм, имеющий квазилинейчатую структуру. Общее содержание О3 определяется многоволновым методом по результатам измерения поглощения атмосферой солнечного излучения на 6 длинах волн 303, 3; 305, 2; 308, 6; 311, 0; 313, 8; 315 нм, совпадающих с максимумами квазилинейчатой структуры. При этом учитывается молекулярное рассеяние и аэрозольное ослабление, которое считается постоянным в узком рабочем спектральном интервале. Рассеянное излучение поглощается светофильтром из кобальтового стекла и кристаллом сульфата никеля. Наземные станции мониторинга озона оснащаются спектрофотометрами Брюэра или Добсона. Спектрофотометр Добсона, хранящийся в Главной геофизической обсерватории им. А.И. Воейкова, является национальным эталоном России. При оптическом наблюдении в верхних слоях атмосферы почти полностью исчезает основное ограничение наземного метода – аэрозольное поглощение. Поэтому озонометры поднимают на большую высоту на шарах-зондах, запускают на метеорологических ракетах и устанавливают на спутниках. Предложен метод наземного дистанционного зондирования озона на миллиметровых радиоволнах (Соломонов С.В., Розанов С.Б., Отделение оптики ФИАН, Россия). Наземное дистанционное зондирование на миллиметровых волнах незаменимо для контроля состояния озонового слоя на высотах от 15 до 75 км и для обнаружения изменений, происходящих под влиянием как динамических, так и химических воздействий, а также для исследования процессов в озонном слое при смене дня и ночи. Этот метод имеет существенные преимущества перед наземными оптическими (УФ-спектрометры и лидары) и контактными (с шаров-зондов, ракет и самолетов) методами. Миллиметровые волны, по сравнению с ИК-, видимым и УФ-излучением, относительно слабо поглощаются в облаках и аэрозолях, поэтому в мм-диапазоне озон можно контролировать круглосуточно и при различных метеоусловиях. Предлагаемый метод предусматривает регистрацию с поверхности Земли собственного теплового излучения озона на частотах одной из вращательных спектральных линий его молекул. Наиболее удобны для измерения лини с центральными частотами 110, 836 и 142, 175 ГГц (длины волн 2, 7 и 2, 1 мм). Эти оптически тонкие линии расположены в окнах прозрачности атмосферы между сильными линиями поглощения кислорода и водяного пара. Говоря о дистанционных методах анализа атмосферного воздуха, следует отметить развивающуюся в настоящее время лазерную спектроскопию. Имеются в виду оптические системы с лучом, проходящим через воздух на расстоянии до нескольких сотен метров (лидары). При зондировании атмосферы лучом лазера аналитический сигнал формируется благодаря избирательному поглощению света теми или иными молекулами. Лидары могут быть размещены на борту самолета. С их помощью можно отыскивать источник выброса того или иного токсиканта, изучать динамику его превращения. Этот подход уже использован для контроля содержания SO2, NO2, CO, O3. Для определения сероводорода был применен прибор «Safeye-424», основанный на дифференциальной оптической абсорбционной спектроскопии в УФ области. Использование луча общей длиной от 30 до 100 м дает весьма низкий предел обнаружения. Однако практическая реализация таких методов встречает ряд трудностей: помехи из-за рассеяния света в результате действия атмосферных факторов, присутствия «неожидаемых веществ»; кроме того, устройства нередко оказываются довольно сложными и дорогими. Точность результатов оказывается невысокой из-за проблемы градуировки приборов.
ОПРЕДЕЛЕНИЕ ФТОРОВОДОРОДА В США, России и других странах в качестве стандартных применяют потенциометрические методики для определения с ионоселективными электродами многих приоритетных загрязнителей воздуха рабочей зоны – аммиака, циановодородной, бромоводородной, фтороводородной и азотной кислот, а также хлороводородной кислоты и газообразных и твердых фторидов. Для определения HF его поглощают из воздуха фильтром, пропитанным K2HPO4, извлекают сконцентрированный на фильтре HF водой и проводят потенциометрическое измерение содержания HF с фторселективным электродом. Измеряемые концентрации 0, 013-1 мг/м3 (ПДК для HF равна 0, 02 мг/м3). Метод достаточно селективный: определению не мешают органические соединения, мешают CFClO CF2O. Источники загрязнения Попадание нефти и её компонентов в ОС вызывает изменение физических, химических и биологических свойств и характеристик природной среды обитания, нарушает ход естественных биохимических процессов. В ходе трансформации углеводородов могут образоваться стойкие к микробиологическому расщеплению ещё более токсичные соединения, обладающие канцерогенными и мутагенными свойствами. Любой из классов НП может стать вредной примесью, загрязняющей воду. В небольших концентрациях нефтяные загрязнения могут влиять на вкус и запах воды, а при больших содержаниях они образуют гигантские нефтяные пятна и становятся причиной экологических катастроф. Последние происходят при разливах нефти или при попадании больших количеств стоков нефтеперерабатывающих или нефтехимических заводов в поверхностные и грунтовые воды. Стоки, попадающие в поверхностные воды, содержат бензин, керосин, топливные и смазочные масла, бензол, толуол, ксилолы, жирные кислоты, фенолы, стероиды, пестициды и металлорганические соединения. Лёгкие НП (например, бензин) частично растворяются в воде, но в основном образуют с водой эмульсии, тяжёлые НП (минеральные масла и смазки) попадают на дно водоёмов и накапливаются в донных осадках. Попадающие в природные воды из различных источников, нефтяные загрязнения имеют тенденцию к рассеиванию и миграции. При этом в поверхностных водах состав НП под влиянием испарения и интенсивного протекания химического и биологического разложения претерпевает за короткий срок быстрые изменения, а в подземных водах, наоборот, процессы разрушения НП заторможены. Главным источником (кроме аварийных разливов) попадания органических соединений нефтяного происхождения в грунтовые воды служат опасные отходы, которые свозятся на промышленные и муниципальные свалки или накапливаются в отстойных прудах и бассейнах. Другим источником загрязнения грунтовых вод НП является утечка горючего из подземных хранилищ. В разных странах в качестве питьевой воды используют воду из поверхностных или подземных источников. К сожалению, все они подвержены загрязнению вредными химическими примесями, в том числе и НП. Канцерогенными для человека и животных являются не только компоненты самой нефти (например, бензол и бенз(а)пирен), но и многочисленные и распространённые в различных сферах деятельности человека продукты нефтехимии (винилхлорид, пестициды, галогенуглеводороды, нитрилы, гидразины и др.). Один из стандартов качества питьевой воды предполагает постоянных контроль за содержанием в питьевой (водопроводной) воде 60 летучих органических соединений (ЛОС) – ароматических углеводородов, относящихся к НП, и хлоруглеводородов, являющихся продуктами нефтехимического производства. Источники загрязнения почвы НП те же, что и в случае воды. Главные из них – разливы нефти и НП, сточные воды и выбросы нефтеперегонных заводов и нефтехимических предприятий, а также вредные отходы химических предприятий, скапливающихся на свалках. В отличии от воды и воздуха, для почвы (и донных осадков) в России не установлены ПДК для суммарного содержания НП. Есть лишь ПДК для бензина (0, 1 мг/л) и некоторых ароматических углеводородов (бензол, кумол, стирол, толуол и ксилолы), которые лежат в диапазоне 0, 1-0, 5 мг/кг. Однако в настоящее время проведена работа по нормированию содержаний НП и нефти в почвах России, результатом которой явилось установление ориентировочно допустимых концентраций (ОДК) этих загрязнителей в почвах. В почвах нефть и НП находятся в следующих формах: · в пористой среде – в парообразном и жидком лекгоподвижном состоянии, в свободной или растворённой водной или водно-эмульсионной фазе · в пористой среде и трещинах – в свободном неподвижном состоянии, играя роль вязкого или твёрдого цемента между частицами и агрегатами почвы, в сорбированном состоянии, связанном на частицах горной породы или почвы, в том числе – гумусовой составляющей почв · в поверхностном слое почвы или грунта в виде плотной органоминеральной массы. Как свободные, так и малоподвижные связанные формы НП отдают летучие фракции в атмосферу, а растворимые соединения – в воду. Со временем этот процесс полностью не прекращается, так как микробиологические процессы трансформации углеводородов приводят частично к образованию летучих и водорастворимых продуктов их метаболизма. Почвы считаются загрязнёнными Н и НП, если их концентрация достигает уровня, при котором: · начинается угнетение или деградация растительного покрова · падает продуктивность сельскохозяйственных земель, нарушается природное равновесие в почвенном биоценозе · происходит вымывание Н и НП из почв в подземные или поверхностные воды · изменяются водно-физические свойства и структура почв · заметно возрастает доля углерода Н и НП в некарбонатном (органическом) углероде почв. Для установления экологически безопасного содержания Н и НП установление нижнего допустимого уровня концентрации недостаточно. Природные экосистемы, в частности почвы, обладают большим потенциалом самоочищения от Н и НП, в них действуют физико-химические и микробиологические процессы разрушения углеводородов нефти. Поэтому, если своевременно установить источник загрязнения, концентрация Н и НП в почве будет снижаться, пока не достигнет безопасного уровня. Важно выявить уровень содержания Н и НП в почвах, выше которого процессы самоочищения резко замедляются и почва сама не может справиться с загрязнением и деградирует. Этот уровень можно назвать верхним допустимым уровнем, или пределом потенциала самоочищения. Почвы, содержащие Н и НП выше верхнего допустимого уровня самостоятельно не выйдут из стадии деградации и будут оказывать устойчивое негативное воздействие на контактирующие с ними компоненты ОС. Естественно, что почвы с таким уровнем загрязнения подлежат рекультивации. ПЕСТИЦИДЫ В ПОЧВЕ Пестициды – собирательное название веществ, используемых в сельском хозяйстве для защиты животных и растений (от слов рestis – зараза, разрушение и сido – убивать). Сюда относят: · гербициды – борьба с сорняками · инсектициды – борьба с вредными насекомыми · зооциды – борьба с грызунами (крысы, мыши, суслики) · фунгициды – борьба с грибковыми болезнями · лимациды – уничтожение моллюсков, слизняков · дефолианты – средства для удаления листьев · десиканты – препараты для высушивание листьев на корню · дефлоранты – вещества для удаления излишних цветов и завязей · реппеленты – для отпугивания насекомых, грызунов
МЕТОДЫ ОПРЕДЕЛЕНИЯ ПЕСТИЦИДОВ Унифицированными методами определения фосфорорганических пестицидов (ФОП) в почве, воде, кормах, лекарственных препаратах являются методы газожидкостной хроматографии и тонкослойной хроматографии. Схема выполнения анализа следующая: ФОП экстрагируют из анализируемых проб ацетоном, CHCl3, дихлорметаном, гексаном
испаряют растворитель (используют вакуумный испаритель
газожидкостная тонкослойная хроматография
пламенно-фотометрическим детектором; носитель – хромосорб, неподвижная жидкая фаза – полифенил- метоксисиликон; температура колонки 175 и 210º С пластинки " Силурол" наносят 0, 01; 0, 05 и 0, 1мл основного стандартного раствора для градуировки. На эту же пластинку наносят 0, 1-0, 2 мл сконцентрированной пробы.
Rf = l/L смесь гексана с ацетоном и ССl4 (в разных cоотношениях) Далее после развития тонкослойной хроматограммы её сушат и проявляют – опрыскивают пластинку различными проявителями: 1. AgNO3. Затем пластинку подставляют под УФ-лампу. Отдельные ФОП проявляются на пластинке в виде черно-серых пятен. 2. PdCl2. На пластинке проявляются желто-коричневые пятна (по-видимому, хелаты палладия с ФОП). 3. Резорциновый проявитель – двухатомный фенол. На пластинке проявляются пятна розово-красного цвета.
Качественной характеристикой различных ФОП является индекс Rf. Например, для подвижной фазы хлороформа этот индекс составляет соответственно: метамидофос – 0, 04; хлорофос – 0, 09; фосфамид – 0, 15; антио – 0, 30; базудин – 0, 32; гетерофос – 0, 33; карбофос – 0, 43; афос – 0, 60; фозалон – 0, 69; метафос – 0, 87; байтекс – 0, 90; фоксим – 0, 93 и т.д. Количественное определение проводят путем сравнения интенсивности окраски и площади пятна с наиболее близким к нему по величине и интенсивности пятном стандарта. Унифицированная методика определения фосфорорганических пестицидов хроматоферментным методом используется для определения остаточных количеств фосфорорганических пестицидов в пищевых продуктах растительного и животного происхождения, лекарственных травах, биосубстратах, почве, воде. Метод основан на экстракции ФОП и их токсичных метаболитов органических растворителем, очистка вымораживанием и на дальнейшем определении тонкослойной хроматографией с ферментным проявлением. Обычно не требуется интенсивной очистки экстрактов, т.к. ферментный ингибиторный тест достаточно чувствителен и специфичен. Фермент – эстераза (используют гомогенизат печени крупного рогатого скота); субстрат – индоксилацетат и др. Препараты, угнетающие эстеразу, например, ФОПы, проявляются на хроматограмме в виде белых пятен на окрашенном фоне. Стандартная российская унифицированная методика определения гербицидов различной химической природы в почве, воде и растительности основана на газохроматографическом определении гербицидов или их модифицированных производных с помощью детектора электронного захвата и термоионного детектора после выделения из образцов ацетоном и очистки экстракта перераспределением в системе двух несмешивающихся жидкостей. При очистке препараты разделяются на группы, в которых отсутствуют соединения, имеющие близкие хроматографические характеристики. Экстракты гербицидов, способных перегоняться с водяным паром, очищают этим методом. Повышение чувствительности метода достигается химическим модифицированием: метилированием свободных карбоновых кислот и бромированием ароматическиз аминов, выделяющихся в результате щелочного гидролиза фениламинов. Таким образом, определение остаточных количеств пестицидов в почве и других матрицах осуществляется после основательной пробоподготовки, включающей экстракцию целевых соединений органическими растворителями с последующей очисткой экстракта методом твердофазной экстракции на модифицированных силикагелях. После очистки пестициды определяют в экстракте методом газовой хроматографии. Фосфорорганические и карбаматные пестициды в объектах окружающей среды, в биопробах определяют также ферментные сенсоры на основе иммобилизованных ферментов. В большинстве случаев речь идет о военных разработках, направленных на определение боевых отравляющих веществ. Для определения токсикантов используют ингибиторные сенсоры, поскольку токсичные вещества имеют, как правило, точно определенные биологические мишени, максимально чувствительные к их действию. Наиболее известны сенсоры на основе холинэстераз – группы ферментов, катализирующих гидролиз сложных эфиров холина:
Поскольку ацетилхолин является одним из наиболее важных и универсальных нейротрансмиттеров, обеспечивающих перенос нервных импульсов, ферменты класса холинэстераз присутствуют в организме всех высших животных. Более того, все ини имеют похожее строение: ацетилхолинэстеразы, выделенные из электрического органа угря и эритроцитов человека, совпадают по аминокислотной последовательности на 70%. Это позволяет использовать в биосенсорах ферменты из различных источников – электрического угря, эритроцитов человека, сыворотки крови лошади. Ингибиторы холинэстеразы, например пестициды, снижают активность фермента, что может привести к судорогам, нарушениям мышечной активности или даже к летальному исходу. Активность фермента снижается также в присутствии ионов тяжелых металлов, ПАВов, азотсодержащих органических соединений. В конце ХХ в. ежегодно фиксировалось до 200000 отравлений фосфорорганическими пестицидами, в основном в странах третьего мира. Интерес к холинэстеразам как компонентам биосенсоров в последнее время усилился в связи с необходимостью противодействия терроризму после использования нервно-паралитических ядов в токийском метро сектой «Аум Сенрике».
Кафедра «Химия»
Нечаева И.А. доцент кафедры «Биотехнология», к.б.н.
КОНСПЕКТ ЛЕКЦИЙ по дисциплине
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА |
Последнее изменение этой страницы: 2019-06-19; Просмотров: 180; Нарушение авторского права страницы














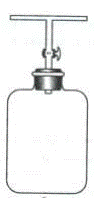














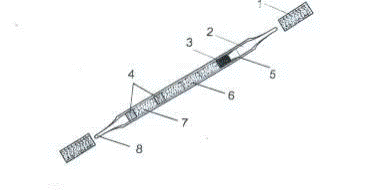



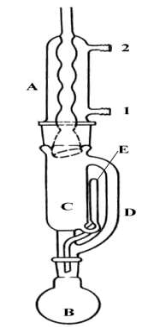


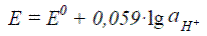






















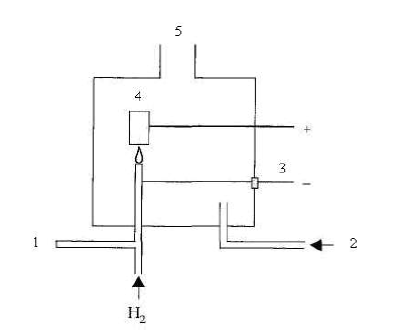


 чтобы избежать разложения ФОП)
чтобы избежать разложения ФОП) хроматография с детектором
хроматография с детектором 
 Подвижный растворитель –
Подвижный растворитель –
