|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Гетероциклические соединенияСтр 1 из 6Следующая ⇒
Введение Сегодня компьютеры стали неотъемлемой частью нашей жизни. Они оказывают неоценимую пользу при подготовке к экзаменам, выполнении домашних заданий, развивают логическое мышление, оказывают помощь в изучении языков и даже дают возможность общаться на дальних расстояниях. В организации системы образования применяются средства с новыми технологиями, однако все еще требуется увеличение количества методических приспособлений. Известно, что все разделы химии в силу свойственных ей особенностей слишком сложны для восприятия. И органическая химия гетероциклических соединений тоже относится к ним. Для лучшего усвоения курса органической химии разработка обучающей и контролирующей программы для студентов ВУЗа, изучающих курс органической химии, является аспектом, позволяющим оптимизацию учебного процесса. Так как в последнее время уделяется особое внимание повышению эффективности учебного процесса, применение компьютера является одним из направлений обучения и тестирования студентов. Электронная версия методической инструкции очень необходима для дистанционного обучения. Электронный учебник обладает всеми особенностями бумажных изданий. К достоинствам электронных учебников относятся: -доступность обращения; -хранение и размещение достаточного количества информации; -возможность осуществлять различные виды учебно–познавательной деятельности (обучать, тестировать и т.д.); -использование механизма гиперссылок дает возможность организовать взаимосвязь между различными понятиями. -интерактивность (наличие обратной связи) достигается за счет включения тестов для самоконтроля; -простота обновления содержания информаций и ее корректировки; -доступность широкому кругу читателей за счет интегрирования в глобальную сеть Internet. Актуальность работы. Внедрение современных информационных технологий, электронного учебника, благодаря которому повышается эффективность изучения данного раздела, активизируется процесс усвоения учебного материала, реализуется индивидуальный подход в обучении. Цель работы: разработать контролирующую программу с обучающим текстом для контроля знаний студентов специальности 5В011200–Химия при изучении раздела «Гетероциклические соединения». Для достижения поставленных целей и решения указанных проблем устанавливаются следующие задачи: - провести анализ теоретического материала, предлагаемого к компьютерной реализации с целью определения его пригодности и степени эффективности; - разработать структуру электронного учебника; - апробация обучающей и контролирующей программы. Научная новизна работы: Впервые разработан электронный учебник по разделу органической химии «Гетероциклические соединения». Практическая значимость заключается в том, что гетероциклические соединения, являясь, основой курса органической химии используется при изучении элективных дисциплин «Биологически активных соединений», «Химия природных соединений», «Химия нуклеиновых кислот», «Гетероциклические соединения». Объект исследования: теоретический и практический материал по данной теме. Дипломная работа выполнена на кафедре химии и биотехнологии Кокшетауского государственного университета им. Ш. Уалиханова.
Пятичленные гетероциклические соединения с одним гетероатомом Фуран
Фуран (оксол–2, 4-диен) — органическое соединение с формулой C4H4O. Пятичленный гетероцикл с одним атомом кислорода. Представляет собой бесцветную жидкость с характерным запахом. Родоначальник большой группы органических соединений, многие из которых имеют практическое значение, например фурфурол, тетрагидрофуран, α -метилфуран (сильван). Фуран был впервые синтезирован в 1870 году. Свойства фурана: Молярная масса=8, 075; Бесцветная горючая жидкость с запахом хлороформа, tкип.31, 330C; tпл.= -85, 60C; d=0, 937; n=1, 4214; уравнения температурной зависимости давления пара: lg p = 8, 4768 - 1671, 4/Т (198-259 К); lg p = 7, 6506-1457, 7/T(276-366 К); ркрит=5, 15МПа; tкрит =2140C; dкрит0, 312 г/см3. Смешивается во всех отношениях с ацетоном, бензолом, СНСl3 и другими органическими растворителями. Слабо растворяется в воде (при 250C в 100 г. воды растворяется 1 г. фурана); в 100 г фурана растворяется 0, 3 г. воды. Образует азеотропные смеси с водой (98, 8% фурана по массе, т. кип. 30, 50C) и 2-метилбутаном (92% фурана, т. кип. 27, 950C). Фуран является ароматическим соединением с шестью p–электронами. Фуран устойчив к действию щелочей, разлагается кислотами.
Фуран – гетероароматическое соединение, свойства которого определяются наличием кольцевого секстета p-электронов, образованных 4 p-электронами двух связей С = С и неподеленной электронной пары гетероатома; другая неподеленная пара электронов атома кислорода остается свободной и может участвовать в образовании оксониевых соединений. Для фурана характерны реакции электрофильного замещенеия, то есть галогенирование, нитрование, сульфирование, ацилирование, меркурирование и т.д. Низкая устойчивость цикла требует проведения этих реакций в " мягких" условиях. Введение электронно-акцепторных заместителей повышает устойчивость ядра фурана; так, например, 2–фуранкарбоновая (пирослизевая) кислота (tпл.1330C, tкип.2300C, 141-144/20 мм рт. ст.) легко нитруется конц. HNO3 в соответствующее 5-нитропроизводное, в то время как фуран в этих условиях разрушается. Реакция фурана с бромцианом приводит к смеси 2-бром- и 2-цианофуранов, формилирование к фурфуролу, озонирование – к смеси глиоксаля и формальдегида, окисление H2O2 в присутствии катализатора Os2O8– к малеиновому ангидриду. Фуран проявляет также свойства диена и может участвовать в диеновом синтезе, например.:
В случае менее активных диенофилов, например акролеина, имеет место заместительное присоединение:
Фуран и его замещенные легко вступают в реакции металлирования. Это используют в препаративных целях, например, для синтеза сульфидов:
Гидрирование фурана над Ni-Ренея (100-160 0C, 16 МПа) приводит к тетрагидрофурану. При более высоких температурах происходит гидрогенолиз фуранового кольца. При действии H2S или NH3(450-500 0C, Al2O3) фуран превращается в тиофен или пиррол (реакция Юрьева). Фуран вместе с 2-метилфураном (сильваном) содержится в продуктах сухой перегонки некоторых пород древесины. В лаборатории фуран обычно получают декарбоксилированием пирослизевой кислоты, в промышленности – декарбонилированием фурфурола:
Для получения тиофена, пиррола, селенофена и малеинового ангидрида, а также в качестве растворителя и экстрагента масел и жиров применяют фуран. Производные фурана используют как лекарственные средства и средства защиты растений. Кислотные свойства фурана усиливаются за счет электронно-акцепторной способности нитрогруппы и карбонильного кислорода. Так, фурацилин и фурадонин относятся к NH–кислотам. Производные фурана как и кислоты взаимодействуют с органическими и неорганическими основаниями. Реакции с разбавленными щелочами приводят к изменению окраски. Для фурадонина характерна прототропная (лактам-лактимная и кетонольная) таутомерия, поэтому взаимодействие с разбавленной щелочью протекает с образованием соответствующих солей. Все рассматриваемые производные фурана образуют продукты разного состава, но близкие по окраске (разные оттенки красного цвета). Поэтому реакция со щелочью используется как общегрупповая при определении подлинности. Такие кислоты Льюиса, как Ag+ , Co2+, Cu2+, Fe2+, образуют окрашенные нерастворимые комплексные соли. Препарат предварительно растворяют в 0, 1 % растворе щелочи, взятой, в строго эквивалентном количестве для переведения веществ в ионное состояние и затем, добавляют 2–3 капли раствора солей. Например, в щелочном спиртовом растворе с ионом меди фурацилин дает осадок темно-красного цвета, фурадонин – болотного цвета, фуразолидон – зеленого цвета. Гидролитическое расщепление. Взаимодействие ЛВ, производных фурана, с концентрированными щелочами приводит к разрыву фуранового цикла и образованию тех или иных продуктов расщепления в зависимости от характера заместителя. Например, фурацилин можно обнаружить по образующемуся продукту разложения – аммиаку. Окислительно-восстановительные свойства ЛВ рассматриваемой группы, содержащие в молекуле гидразиновую группу =N—NH—, обладают восстановительными свойствами. Поэтому для их количественного определения может быть использована йодометрия. Для этого ЛВ, например фурацилин, растворяют в воде с добавлением NаСl, нагревают, далее добавляют избыток титрованного 0, 01М раствора йода и 0, 1 мл 10 % раствора щелочи, затем выдерживают 3–4 мин и подкисляют серной кислотой. Избыток йода оттитровывают раствором Na2S2O3:
Испытания на чистоту. Смешать 1, 0 г ЛВ со 100 мл, встряхнуть и отфильтровать. Фильтр должен иметь рН 5, 0–7, 0. Родственные примеси, например нитрофуран, определяют методом ЖХ. Потеря массы при высушивании–не более 0, 5 % при навеске 1, 000 г (Г=100-105 °С); сульфатная зола – не более 0, 1 %. Количественное определение. Проводят методом УФ-спектрофотометрии: готовят раствор, как описано при определении подлинности (в защищенном от яркого света месте). При приготовлении раствора используют стандартный образец нитрофурала. Измерение абсорбции проводят для обоих растворов. Тиофен Тиофен– органическое соединение с формулой C4H4S. Подобно бензолу, тиофен является представителем большой группы соединений. По физическим и химическим свойствам тиофен весьма сходен с бензолом, а производные тиофена, как по способам образования, так и по другим свойствам близко стоят к соответственным производным бензола. Бензол и его производные, получаемые из природных продуктов, обыкновенно сопровождаются примесями тиофена или соответственных производных группы тиофена. Открыт тиофен был Виктором Мейером в 1883 году в бензоле, полученном из каменного дегтя. В. Мейер показал, что прежде приписываемая бензолу реакция окрашивания в синий цвет от прибавки изатина и серной кислоты происходит на счет примеси к бензолу тиофена в количестве около 0, 5%. Бензол, полученный перегонкой соли бензойной кислоты, не обнаруживает реакции с изатином. Выделение тиофена из раствора его в бензоле основано на более легкой способности тиофена, реагировать с серной кислотой. Для выделения и очищения тиофена, сульфосоединение переводится в свинцовую соль действием углекислого свинца. Свинцовую соль сульфотиофена подвергают сухой перегонке с эквивалентным количеством нашатыря. Дистиллят растворяют в 100 объемов лигроина(предварительно очистив последний взбалтыванием с дымящей серной кислотой) и взбалтывают лигроинный раствор с крепкой серной кислотой в продолжении двух часов (10 объемов кислоты на 1 объем дистиллята). Затем кислый слой сразу разбавляют водой, нейтрализуют углекислым свинцом и свинцовую соль сульфотиофена снова разлагают, как уже описано выше. Полученный таким образом тиофен фракционируют. Для того чтобы получить тиофен без примеси бензола нужно взбалтывать бензол из каменноугольного дегтя с малым количеством крепкой серной кислотоы, напиример четыре части кислоты на сто частей бензола. Вместо усреднения сульфосоединения и разложения соли можно разбавлять сульфосоединение двумя или тремя объемами воды и отгонять тиофен водяным паром. Синтетически тиофен можно получить при пропускании сернистого этилена или ацетилена в кипящую серу; при пропускании сернистого этила через накаленную трубку; при пропускании этилена, светильного газа или паров лигроина через накаленный пирит; при кипячении кротоновой, нормальной масляной кислоты или паральдегида с сернистым фосфором; действием сернистого фосфора на эритрит. Лучше всего получать синтетический тиофен из ангидрида янтарной кислоты действием трехсернистого фосфора на натровую соль янтарной кислоты (Фольхардт и Эрдман). Тиофен представляет собой бесцветную жидкость, которая имеет слабый запах и кипит при температуре 84 и застывающая в охладительной смеси из твердой угольной кислоты и эфира. Удельный вес тиофена равен 1, 08844. Молекулярная теплота горения составляет 669, 5 калорий. Большинство химических реакций, характеризующих бензол, протекают также аналогично идля тиофена. Так хлор, бром, йод, азотная кислота и серная кислота действуют на тиофен, образуя галоидозамещенные нитросоединения и сульфосоединения тиофена. При кипячении с натрием тиофен не изменяется. Действиемнатрия на йодтиофен и йодистый метил образуется метилтиофен C4H3SCH3. При окислении метилтиофена образуется тиофенкарбоновая кислота C4H3SCOOH, аналогичная бензойной кислоте. Нитротиофен C4H3SNO2 восстанавливается в аминотиофен C4H3SNH2. При кипячении трифенилкарбинола с тиофеном образуется трифенилтиенилметан (C6H5)3CC4H3S( остаток тиофена C4H3S аналогичный фенилу при бензоле называется тиенил). Подобно бензолу, тиофен склонен к уплотнениям, например с бензойным альдегидом при действии фосфорного ангидрида тиофен образует дитиенилфенилметан (C4H3S)2CHC6H5. С хлористым бензоилом в присутствии хлористого алюминия из тиофена получается тиенилфенилкетон C4H3S—CO—C6H5. При стоянии тиофена с насыщенным водным раствором сулемы в присутствии уксуснокислого натрия и спирта образуются в виде осадков соединения C4H3SHgCl и C4H2S(HgCl)2. При кипячении с водой первое соединение переходит в раствор. При действии концентрированной хлористоводородной кислоты этиртутные соединения тиофена разлагаются на сулему и тиофен. С хлорангидридами кислот они с легкостью образуют кетоны типа C4H3S—CO—R. С раствором сернокислой ртути тиофен образует осадок. Эта реакция служит для количественного определения тиофена в бензоле. Кроме упомянутой уже ранее индофенинной реакции (изатин и серная кислота), тиофен можно характеризовать еще несколькими цветными реакциями, так например, с небольшим количеством азотистокислого эфира изоамилового спирта в присутствии серной кислоты происходит темно-красное, затем переходящее в темно–фиолетовое окрашивание. С нитрозосерной кислотой зеленое окрашивание переходит в синее окрашивание. С веществами типа R—CO—CO—R' в присутствии серной кислоты тиофена дает окрашенные продукты уплотнения. Аналогия бензола тиофена в реакциях дала повод к заключению о строении тиофена, подобном строению бензола. Вследствии аналогичности бензолу, Виктор Мейер принял строение тиофена циклическим с заменой в бензоле двуэквивалентной группы —СН=СН— двуэквивалентным атомом серы:
Такое строение тиофена подтверждается способностью его образовавать два ряда однозамещенных производных α и β, четыре ряда двузамещенных производных при одинаковых заместителях (один α α, один β β и два α β ) и т.д. В пользу указанного строения тиофена можно привести еще образование как самого тиофена, так и его производных при действии сернистых соединений фосфора на вещества с открытой углеродной цепью, имеющие карбонильные группы в положении 1, 4, т.е. – СО-С-СО, например янтарная кислота, левулиновая кислота, ацетонилацетон и др. Итак, тиофен нужно отнести к гетероциклическим соединениям, т.е. к соединениям с замкнутой цепью, звеньями которой являются разнородные атомы. Номенклатура производных в группе тиофена построена так же как в производных группы бензола. Метилтиофен называется тиотоленом, диметилтиофен называется тиоксеном и т.д. Производные тиофена являются спутниками производных бензола, например, тиотолен и тиоксен мжно выделить соответственно из толуола и ксилола, полученных из каменноугольного дегтя. Основанием для определения ряда однозамещенных производных α -или β служат синтетические реакции, например.: из метилянтарной кислоты получается β –тиотолен, а из левулиновой α -тиотолен:
Из ацетонилацетона получается α α –тиоксен, а из метиллевулиновой кислоты α β -тиоксен. Получен также тилфен, пропилфен, бутилфен и октилфен. Все четыре возможные по теории изомерные тиоксены. Также получены триметилтиофен из диметиллевулиновой кислоты. Фенилтиофен C6H5—C4H3S получен из бензоилпропионовой кислоты. При пропускании тиофена через накаленную трубку образуется дитиенил C4H3S—C4H3S, аналогичный дифенилу. Продукты замещения образуются у тиофена легче, чем у бензола. Так, если в тиофене имеется боковая цепь, то заместить водород боковой цепи галоидами нельзя. Водороды тиофенного кольца всегда замещаются, а в ряде бензола при высокой температуре замещается водород боковой цепи. При пропускании в тиофен хлора можно получить монохлортиофен, дихлортиофен, трихлортиофен и тетрахлортиофен. Характерные реакции окрашивания для тиофена повторяются и в его производных. При действии едкого калии спирта динитротиофен дает интенсивное окрашивание ярко красного цвета. Реакция настолько чувствительна, что можнл обнаружить 0, 0000001 г динитротиофена в двух кубических сантиметрах раствора. Из аминотиофена, называемого аналогично анилину тиофенином, получены азокраски. Действием азотистой кислоты на тиофенин получен тиенол C4H3SOH аналог фенола. Известны также тиениловый спирт C4H3SCH2OH, альдегид C4H3SCHO и кислота C4H3SCOOH. Аналог нафталина, тиофен, получен действием сернистого фосфора на лимонную кислоту:
Итак, несмотря на существенную разницу по химическому составу соединений тиофеновой бензольной групп, можно наблюдать полное сходство, как типов соединений, так и превращений веществ в обеих группах. Это дает право сделать заключение, что в некоторых случаях свойства и превращения органических веществ зависят от группировки атомов в частице в гораздо большей степени, чем от химического состава соединения.
Пиррол Пиррол С4H4NН – пятичленный гетероцикл с одним атомом азота.
Бесцветная жидкость с температурой кипения 130°С, плохо растворимая в воде, на воздухе быстро окисляется и темнеет. Электронное строение молекулы пиррола объясняет его свойства как слабой кислоты и ароматического соединения.
Атомы углерода и азота находятся в состоянии sp2–гибридизации.
Устойчивость пиррола как ароматической структуры значительно меньше, чем бензола. Под действием сильных минеральных кислот электронная пара азота все же используется для солеобразования и свойства пиррола резко меняются: ароматичность исчезает (в системе сопряжения остается всего 4 электрона) и проявляются свойства диена, например, способность к полимеризации. Связывание неподеленной электронной пары атома азота системой сопряжения приводит к резкому ослаблению основных и проявлению кислотных свойств. Как слабая кислота пиррол вступает в реакцию с металлическим калием, образуя соль – пиррол–калий:
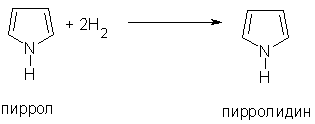
Пиррол может участвовать в реакциях присоединения: гидрирование приводит к образованию пирролидина
под действием сильных минеральных кислот пиррол вступает в реакции полимеризации. Пиррол применяют для синтеза различных органических веществ. Пиррольные структуры содержатся в гемоглобине, хлорофилле, витамине В12 и некоторых других природных соединениях. В состав молекул этих сложных веществ входит тетрапиррольный фрагмент (порфин) в виде комплекса с металлом:
где, Ме – металл (Fe в гемоглобине, Mg в хлорофилле, Co в витамине В12). Химические свойства 1.Сильные минеральные кислоты могут вытягивать электронную пару атома азота из ароматической системы, при этом ароматичность нарушается, и пиррол превращается в неустойчивое соединение, которое сразу полимеризуется. Неустойчивость пиррола в кислой среде называется «ацилофобностью». 2.Пиррол проявляет свойства очень слабой кислоты. Он реагирует с калием, образуя пирролкалий:
3.Пиррол как ароматическое соединение склонен к реакциям электрофильного замещения, которые протекают преимущественно у α -атома углерода (соседнего с атомом азота). Поскольку пиррол под действием кислот полимеризуется, то для проведения замещения используют реагенты, не содержащие протонов. Так, для нитрования пиррол а используют ацетилнитрат:
а для сульфирования – комплекс пиридина с оксидом серы (VI):
4.При гидрировании пиррола образуется пирролидин – циклический вторичный амин, проявляющий значительные основные свойства.
Интересными свойствами обладают пятичленные ароматические гетероциклы, содержащие два атома азота, имидазол и пиразол C3H4N2:
Эти соединения амфотерны. Один атом азота (пиридинового типа) в них проявляет слабые основные свойства и способен принимать протон, а другой атом азота (пиррольного типа) входит в состав группы NH, которая проявляет слабые кислотные свойства и способна отдавать протон. Благодаря этому появляется возможность переноса протона между двумя атомами азота, и некоторые производные имидазола и пиразола могут существовать в виде быстро переходящих друг в друга таутомерных форм:
Индол Индол (бензопиррол, 2, 3-бензпиррол). Внешний вид: бесцветные листовидные кристаллы Брутто – формула: C8H7N Молекулярная масса: 117, 15 Бесцветное кристаллическое вещество с запахом нафталина, растворимое в горячей воде; температура плавления 52°C. Температура кипения: 254. Бесцветные кристаллы с запахом, напоминающим запах капусты. Индольное ядро входит во множество биохимических природных соединений. Как индивидуальное вещество содержится в каменноугольной смоле, и в некоторых эфирных маслах, например, в масле жасмина. Бесцветные листовидные кристаллы с запахом гнилой капустной кочерыжки. Химические свойства индола аналогичны свойствам пиррола.
Пиразол Номенклатура производных пиразола основана на принятой для производных пиррола. Нумерацию начинают с атома азота иминогруппы и продолжают в направлении второго атома азота. Пиразол (кристаллическое вещество, т.пл.70°C) и его производные найдены в природе. Они получаются синтетически и находят широкое применение в качестве лекарственных препаратов и красителей. Пиразол образуется при действии диазометана на ацетилен:
HC CH HC - CH
HC N HC N \ / N NH Химические свойства.1. Пиразол является основанием. Он устойчив к действию окислителей, а также кислот и щелочей. В противоположность пирролу он нитруется и сульфируется соответствующими кислотами в положение 4, т.е. не проявляет ацидофобных свойств.4-Аминопиразол получается восстановлением 4–нитропроизводного, он способен диазотироваться и сочетаться с солями диазония: HC CH H2SO4 || + || + HNO3 —→ O2N – C – CH + H2O HC N || || \ / HC N NH \ / NH | [H] |4-нитропиразол Н2N – C – CH || || HC N \ / NH 4-аминопиразол Водородом в момент выделения (натрий и спирт) пиразол медленно восстанавливается в дегидропроизводное пиразолин: HC—CH HC — CH Н2C—СН2 || || | || | | HC N HC N H2C NH \ / \ / \ / NH NH NH пиразол пиразолин пиразолидин Пиразолин - более сильное основание, чем пиразол. Легко окисляется. Пиразолон и его производные.Пиразолону отвечают важные производные группы пиразолона: H2C—CH | || OC N \ / NH Производные пиразолона легко получаются конденсацией замещенных гидразинов с эфирами ß -кетонокислот или присоединением к дикетену. При конденсации фенилгидразина с ацетоуксусным эфиром с отщеплением воды и спирта образуется 1–фенил–3метилпиразолон–5 (Кнорр, 1883г.) Имидазол Имидазол– органическое соединение класса гетероциклов, пятичленный цикл с двумя атомами азота и тремя атомамиуглерода в цикле, изомерен пиразолу. В незамещенном имидазоле положения 4 и 5 (атомы углерода) равноценны, вследствие таутомерии. Ароматичен, реагирует с солями диазония (сочетание).Нитруется и сульфируется только в кислой среде в положение 4, галогены в щелочной среде вступают по положению 2, в кислой - по положению 4.Легко алкилируется и ацилируется по иминному N, раскрывает цикл при взаимодействии с растворами сильных кислот и пероксидов.Катализирует гидролиз трудноомыляемых сложных эфиров и амидов карбоновых кислот. На основе имидазола производят большое количество различных ионных жидкостей. Впервые имидазол был получен Генрихом Дебюсом в 1858 г. конденсацией глиоксаля с аммиаком и формальдегидом, этот метод может использоваться и в синтезе замещенных имидазолов:
В лабораторной практике имидазол синтезируют декарбоксилированием 4, 5-имидазолдикарбоновой кислоты, получаемой окислением бензимидазола.
Тиазол Тиазол– (температура кипения 117°C) гетероциклическое соединение, аналог тиофена, содержащий вместо CH-группы в положении 3 атом азота. Тиазол представляет собой бесцветную жидкость с запахом пиридина. Тиазол растворим в воде и в органических растворителях. Как и молекулы других азолов, молекула тиазола обладает гетероароматической 6π -электронной системой. По своим химическим свойствам тиазол приближается к пиридину и тиофену. Тиазол вступает в реакции электрофильного замещения, но его реакционноспособность снижается вследствие N-протонирования или комплексообразования с кислотами Льюиса. Однако в отсутствие кислот Льюиса реакции электрофильного замещения, в частности, нитрование в среде уксусного ангидрида и бромирование в бензоле, идут в положение 5 по механизму образования σ -комплекса. При этом реакционноспособность уменьшается в ряду 5- > 2- > 4-положение. Также возможно протекание реакций по илидному механизму, например, реакция изотопного обмена:
Тиазол вступает также в реакции нуклеофильного замещения. Легче всего замещение идёт в положение 2 по механизму реакции Чичибабина, например, замещение атома водорода на аминогруппу и реакции металлирования. С образованием N-оксидов по атому азота происходит образование тиазола. Тиазол устойчив к восстановителям, например к водороду в момент выделения и в присутствии катализаторов. В то же время он реагирует с сильными восстановителями: никель Ренея разрушает молекулу тиазола с десульфуризацией и образованием алифатических соединений, а борогидрид натрия NaBH4 превращает его в тетрагидротиазол. Основные методы синтеза тиазола и его замещенных: взаимодействие a-галогенкетонов или a-галогенальдегидов с тиоамидами, циклизация a-тиоцианокетонов под действием водных растворов кислот или безводного НСl либо РОСl3, взаимодейтвие ациламинокарбонильных соед. с P2S5, напр.:
Синтез тиазола и его производных осуществляется из аминотиолов и из тиоамидов по общей схеме:
Некоторые производные тиазола являются лекарственными препаратами: 2-сульфаниламидотиазол (норсульфазол) и 2-(N-о-карбоксибензоилсульфанил) амидотиазол (фталазол). Тиазолидиновый цикл является структурными фрагментом молекулы пенициллина и его полусинтетических аналогов. Цикл тиазола входит в составтиамина. Некоторые окрашенные производные тиазола являются красителями. Пиридин Пиридин– шестичленный ароматический гетероцикл с одним атомом азота, бесцветная жидкость с резким неприятным запахом; смешивается с водой и органическими растворителями. Пиридин – слабое основание, дает соли с сильными минеральными кислотами, легко образует двойные соли и комплексные соединения. Пиридин был известен ещё алхимикам, но первое письменное описание этого вещества было сделано шотландским химиком Томасом Андерсоном в 1851 году. Он обнаружил его при исследовании костяного масла, получающегося сухой перегонкой необезжиренных костей, среди прочих веществ, была получена бесцветная жидкость с неприятным запахом. В 1869 году Кернер в частном письме к Каниццаро высказал мысль, что пиридин, может быть рассматриваем, как бензол, в котором одна группа СН замещена азотом. По мнению Кернера, подобная формула не только объясняет синтезы пиридина, но, главным образом, указывает, почему простейший член ряда пиридиновых оснований имеет пять атомов углерода. Через год Дьюар, независимо от Кернера, пришел к той же формуле, которая затем нашла себе подтверждение и в позднейших работах других химиков. Позже изучением структуры пиридина занимались Томсен, Бамбергер и Пехманн, Чамичан и Деннштедт. В 1879 году А. Вышнеградский высказал мнение, что, может быть, все растительные основания являются производными пиридина или хинолина, а в 1880 году Кенигс предлагал даже именем алкалоидов называть только те растительные основания, которые могут быть рассматриваемы, как производное пиридина. Однако на настоящее время границы понятия «алкалоиды» значительно расширились. Основным источником для получения пиридина является каменноугольная смола. Пиридин проявляет свойства, характерные для третичных аминов: образует N-оксиды, соли N-алкилпиридиния, способен выступать в качестве сигма-донорного лиганда. В то же время пиридин обладает явными ароматическими свойствами. Однако наличие в кольце сопряжения атома азота приводит к серьёзному перераспределению электронной плотности, что приводит к сильному снижению активности пиридина в реакциях электрофильного ароматического замещения по сравнению с бензолом. В таких реакциях реагируют преимущественно мета-положения кольца. Для пиридина характерны реакции ароматического нуклеофильного замещения, протекающие преимущественно по 2- или 4- положениям кольца. Такая реакционная способность свидетельствует о электронно-дефицитной природе пиридинового кольца, что может быть обобщено в следующем эмпирическом правиле: реакционная способность пиридина как ароматического соединения примерно соответствует реакционной способности нитробензола. Применяют в синтезе красителей, лекарственных веществ, инсектицидов, в аналитической химии, как растворитель многих органических и некоторых неорганических веществ, для денатурирования спирта.
Пиримидин Пиримидин(C4N2H4, 1, 3- или м-диазин, миазин) – гетероциклическое соединение, имеющее плоскую молекулу, простейший представитель 1, 3-диазинов. Пиримидин– бесцветные кристаллы с характерным запахом. Молекулярная масса пиримидина 80, 09 г/моль. Пиримидин проявляет свойства слабого двукислотного основания, так как атомы азота могут присоединять протоны за счет донорно-акцепторной связи, приобретая при этом положительный заряд. Реакционная способность в реакциях электрофильного замещения у пиримидина снижена из-за снижения электронной плотности в положениях 2, 4, 6, вызванного наличием двух атомов азота в цикле. Так, пиримидин не нитруется и несульфируется, однако в виде соли бромируется в положение 5. Электрофильное замещение становится возможным только при наличии электронодонорных заместителей и направляется в наименее дезактивированное положение 5. Под действием алкилирующих агентов (алкилгалогениды, борфторид триэтилоксония) пиримидин образует четвертичные N-пиридиниевые соли, при действии перекиси водорода и надкислот образует N-оксид. Реакции пиридина с азотными нуклеофилами зачастую сопровождаются раскрытием кольца с дальнейшей рециклизацией: так, в жестких условиях при взаимодействии с гидразином, пиримидин образует пиразол, при взаимодействии с метиламином— 3-этил-5-метилпиридин. Пониженная электронная плотность кольца приводит к тому, что пиримидин активен по отношению к нуклеофильным реагентам, которые атакуют 2-, 4- и 6- атомы углерода цикла. Так, литийорганические соединения и реактивы Гриньяра присоединяются к пиримидину с образованием 4-замещенных 3, 4-дигидропиримидинов Пиримидин получают восстановлением галогенизированых пиримидиновых производных. Или из 2, 4, 6-трихлор пиримидина, получаемого обработкой барбитуровой кислоты хлороксидом фосфора. Производные пиримидина широко распространены в живой природе, где участвуют во многих важных биологических процессах. В частности, такие производные как цитозин, тимин, урацил входят в состав нуклеотидов, являющихся структурными единицами нуклеиновых кислот, пиримидиновое ядро входит в состав некоторых витаминов группы B, в частности B1, коферментов и антибиотиков.
· Структурная формулацитозина
· Структурная формулатимина
· Структурная формулаурацила Пиримидиновая структура — как ароматическая, так и гидрированная, входит в состав многих биологически активных веществ и лекарственных препаратов — например, барбитуратов – производных 1, 3, 5–тригидроксипиридина, обладающих снотворным, противосудорожным и наркотическим действием.
Способы получения Методы получения пятичленных гетероциклов Пиррол и тиофен содержатся в каменноугольной смоле. Фракционной перегонкой смолы тиофен (Тпл 84оС) перегоняется вместе с бензолом (Ткип 80оС) и содержание его в бензоле 0, 5% (1884 г., В.Мейер). Тиофен в промышленности может быть получен при взаимодействии бутана с серой при 560 оС:
Популярное:
|
Последнее изменение этой страницы: 2016-07-12; Просмотров: 2337; Нарушение авторского права страницы