
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Предмет и методы химической термодинамики. Взаимосвязь между процессами обмена веществ и энергии в организме. Химическая термодинамика как теоретическая основа биоэнергетики.Стр 1 из 10Следующая ⇒
Предмет и методы химической термодинамики. Взаимосвязь между процессами обмена веществ и энергии в организме. Химическая термодинамика как теоретическая основа биоэнергетики. Термодинамика - это раздел физической химии, изучающий взаимные превращения различных видов энергии, связанные с переходом энергии в форме теплоты и работы. Объектом термодинамического исследования есть термодинамическая система. Для описания явлений, происходящих в термодинамических системах, используется ряд понятий: 1. термодинамическая система – тело или совокупность тел, обособленных физическими или воображаемыми границами от окружающей среды; 2. фаза – часть системы, обладающая одинаковым составом, физическими и химическими свойствами, имеющая поверхность раздела, отделяющую ее от других частей; 3. термодинамический процесс – изменения в термодинамической системе.
Системой называют совокупность объектов отделенных из окружающего мира реально существующими или воображаемыми поверхностями. Системой может быть газ в сосуде, раствор реагентов в колбе, кристалл вещества или даже мысленно выделенная часть этих объектов. По взаимодействию с окружающей средой термодинамические системы делят на: открытые – обмениваются с окружающей средой веществом и энергией (например, живые объекты); •закрытые – обмениваются только энергией (например, реакция в закрытой колбе или колбе с обратным холодильником), наиболее частый объект химической термодинамики; •изолированные – не обмениваются ни веществом, ни энергией и сохраняют постоянный объем (приближение – реакция в термостате). В основе биоэнергетики организмов лежат законы термодинамики, одинаковые для живых и неживых систем. В соответствии с ее законами живой организм представляет собой открытую стационарную неравновесную систему, обменивающуюся с окружающей средой веществом и энергией, постоянство параметров которой обеспечивается непрерывным поступлением энергии из окружающей среды в количестве, компенсирующем его внутренние расходы. Основные понятия термодинамики. Интенсивные и экстенсивные параметры. Функция состояния. Внутренняя энергия. Работа и теплота - две формы передачи энергии. Физические свойства, характеризующие состояние системы, называют параметрами состояния системы. Взаимодействие системы с окружающей средой заметно по изменению параметров системы. Интенсивные параметры – это параметры, которые не зависят от количества вещества и выравниваются при объединении систем (температура, давление, концентрация, плотность, поверхностное натяжение). Параметры состояния связаны уравнением состояния. Экстенсивные параметры – это параметры, которые зависят от количества вещества системы и суммируются при объединении систем (объём, масса, энергия, площадь и т.д.). Функция состояния - это характеристика системы, которая не поддается прямому измерению, а рассчитывается через параметры состояния. Значение функции состояния не зависит от способа его достижения, а только от начального и конечного состояния системы. К ним относятся, например, давление, объем, температура системы. Каждая термодинамическая система обладает определенным запасом энергии, которая называется внутренней энергией. Внутренняя энергия U – это общий запас энергии системы, слагающийся из кинетической энергии движения составляющих ее частиц (молекул, атомов, ионов, электронов и др.) и потенциальной энергии их взаимодействия. Внутренняя энергия системы есть функцией ее состояния и зависит от параметров системы. Величина внутренней энергии зависит от природы тела, его массы, химического состава и параметров, которые обусловливают состояние системы – давления, объема, температуры. Для термодинамического анализа достаточно знать только прирост внутренней энергии Δ U = U конеч - U начал Энтальпия - это энергия, которой владеет система при постоянном давлении. H = U + pV
pV – потенциальная энергия Энтальпия имеет большое значение в химии, так как передача тепла в химической реакции происходит при постоянном давлении. Работа (А) – это форма передачи энергии, вследствие чего система развивает напрямленую силу и делает работу над другой системой, к которой эта сила приложена. Работу которую делает система на окружающей средой считают положительной А> 0 (+А), а работу которая делается над системой – отрицательной А< 0 (-А). Теплотой (Q) называют форму передачи энергии от одной системы к другой вследствие неналаженого (хаотического) движения молекул. В химической термодинамике различают процессы: Изохорные - происходящие при постоянном объеме (V=const) Изобарные - происходящие при постоянном давлении (р=const) Изотермические- происходящие при постоянной температуре (Т=const) Адиабатические- происходящие без обмена тепла с окружающей средой, система не получает тепла извне и не отдает его окружающей среде (Q = 0) Типы термодинамических систем (изолированные, закрытые, открытые). Типы термодинамических процессов (изотермические, изобарные, изохорные). Стандартное состояние. По взаимодействию с окружающей средой термодинамические системы делят на: открытые – обмениваются с окружающей средой веществом и энергией (например, живые объекты); •закрытые – обмениваются только энергией (например, реакция в закрытой колбе или колбе с обратным холодильником), наиболее частый объект химической термодинамики; •изолированные – не обмениваются ни веществом, ни энергией и сохраняют постоянный объем (приближение – реакция в термостате). В химической термодинамике различают процессы: Изохорные - происходящие при постоянном объеме (V=const) Изобарные - происходящие при постоянном давлении (р=const) Изотермические- происходящие при постоянной температуре (Т=const) Состояние системы определяется совокупностью ее физических и химических свойств. Каждое состояние системы характеризуется определенными величинами этих свойств. Если эти свойства изменяются, то изменяется и состояние системы, если же свойства системы не изменяются со временем, то система находится в состоянии равновесия. Для сравнения свойств термодинамических систем необходимо точно указать их состояние. С этой целью введено понятие – стандартное состояние, за которое для индивидуальной жидкости или твердого тела принимается такое физическое состояние, в котором они существуют при давлении в 1 атм (101315 Па) и данной температуре 298К. Величины, относящиеся к стандартному состоянию, пишутся с индексом «о». Так, например, уравнение Клапейрона–Менделеева является уравнением состояния идеального газа: РV = nRT где Р – давление, V – объем, n – число молей идеального газа, Т – его абсолютная температура и R– универсальная газовая постоянная. Следствия из закона Гесса 1) Тепловой эффект кругового процесса равен нулю. Круговой процесс - система, выйдя из начального состояния, в него же и возвращается. DH1+DH2-DH3= 0 Отсюдаа же вытекает и закон Лавуазье-Лапласа. 2) Тепловой эффект реакции равен сумме теплот образования продуктов реакции за вычетом суммы теплот образования начальных (исходных) веществ. 3) Тепловой эффект реакции равен сумме теплот сгорания исходных веществ за вычетом суммы теплот сгорания конечных продуктов. Термохимические процессы: Экзотермические – реакции, при протекании которых происходит уменьшение энтальпии системы (Δ Н < 0) и во внешнюю среду выделяется теплота Эндотермические – реакции, в результате которых энтальпия возрастает (Δ Н > 0) и система поглощает теплотуQизвне. 6.Второе начало термодинамики. Формулировка. Обратимые и необратимые в термодинамическом смысле процессы. Энтропия как критерий возможности протекания самопроизвольных процессов Второе начало термодинамики: В изобарно-изотермических условиях (р, Т =const) в системе самопроизвольно могут протекать только такие процессы, в результате которых энергия Гиббса системы уменьшается (Δ G< 0). В состоянии равновесияG=const, G= 0 Процессы могут быть обратимые и необратимые. Термодинамически обратимым называется процесс, который можно реализовать в прямом и обратном направлениях при этом система возвращается в исходное состояние через промежуточные состояния равновесия не оставляя изменений в окружающей среде. Необратимыми называют процессы, при которых в результате прямого и следующего за ним обратного перехода в системе или окружающей среде возникают какие либо неисчезающие изменения. Все реальные самопроизвольно идущие процессы – необратимы. В реальных необратимых системах только часть энергии превращается в полезную работу, другая часть является как бы связанной, «обесцененной». Для характеристики этой энергии используют функцию энтропию S. Δ Q Δ S =---------------------------- T Смена энтропии Δ S определяется только начальным и конечным станами системы: Δ S = Sконеч - Sначал Энтропия есть мерой рассеянной (обесцененной) энергии. Чем больше величина энтропии тем меньшая часть энергии может превратится в работу, то есть энтропия выступает как мера необратимости процесса. Энтропия – мера вероятности пребывания системы в данном состоянии – мера неупорядоченности системы. Энергия Гиббса – главный критерий возможности протекания самопроизвольных про- цессов. Прогнозирование направления самопроизвольно протекающих процессов в изолированной и закрытой системах; роль энтальпийного и энтропийного факторов. Энергия Гиббса – функция состояния, являющаяся критерием самопроизвольности процессов в открытых и закрытых системах. Δ G=Δ H–TΔ S cd(D)ce(E) Δ rG=Δ rG0+RTlnca(A)cb(B) – изотерма Вант-Гоффа Критериями направления самопроизвольного протекания необратимых процессов являются неравенстваΔ G< 0 (для закрытых систем), Δ S> 0 (для изолированных систем) В ходе самопроизвольного процесса в закрытых системах Gуменьшается до определенной величины, принимая минимально возможное для данной системы значениеGmin. Система переходит в состояние химического равновесия (Δ G= 0).
Самопроизвольное течение реакций в закрытых системах контролируется как энтальпийным (Δ rH ), так и энтропийным (TΔ rS) фактором. Для реакций, у которых Δ rH< 0 иΔ rS> 0, энергия Гиббса всегда будет убывать, т.е.Δ rG< 0, и такие реакции могут протекать самопроизвольно при любых температурах В изолированных системах энтропия максимально возможное для данной системы значениеSmax; в состоянии равновесияΔ S= 0
8. Термодинамические условия равновесия. Стандартная энергия Гиббса образования вещества, стандартная энергия Гиббса биологического окисления вещества. Стандартная энергия Гиббса реакции. Примеры экзергонических и эндергонических процессов, протекающих в организме. Принцип энергетического сопряжения. Изобарный и изохорный потенциалы есть функциями состояния системы. Их используют для определения направления пути процесса при условии термодинамического равновесия. Для расчетов используют Δ G и Δ F. Если Δ G и Δ F равны нулю, то система находится в состоянии равновесия. Когда Δ G 0 и Δ F 0, то процесс может идти самопроизвольно с преобразованием энергии в полезную работу. В случае если Δ G 0 и Δ F 0, то изменение состояния системы происходит только при использовании внешней работы. Условием самопроизвольного пути химических процессов есть повышение энтропии и уменьшение энергии Гиббса, а условием термодинамического равновесия есть максимальное значение энтропии и минимальное значение энергии Гиббса.
Стандартная энергия Гиббса: Δ rG = Σ υ jΔ jG0j – Σ υ iΔ iG0i Экзергонические реакции – G < 0 и системой совершается работа (окисление глюкозы) Эндергонические – G > 0 и над системой совершается работа. В живых системах эндергонические реакции происходят за счет сопряжения с экзергоническими. Это возможно, если обе реакции имеют какое-либо общее промежуточное соединение. Энергетическое сопряжение. В сопряженной системе определяющим фактором будет сумма разностей потенциалов двух процессов (Δ РЭФФ = Δ P1 + Δ P2). Суммарный процесс возможен при условии, если Δ РЭФФ — величина отрицательная. Благодаря энергетическому сопряжению возможно взаимопревращение одних форм работы и энергии в другие. Общая константа последовательно и параллельно протекающих процессов. Уравнения изотермы и изобары химической реакции. Прогнозирование смещения химического равновесия. Понятие о буферном действии, гомеостазе и стационарном состоянии живого организма. Пусть некоторая реакция описывается уравнением: aA + bB = cC + dD · a, b, c, d - коэффициенты уравнения реакции; · A, B, C, D - химические формулы веществ. Константа равновесия: [C]c[D]d K = ———————— [A]a[B]b Уравнение изотермы химической реакции: Δ Gр-я = RTln(Π c/Kc) Химическая кинетика как основа для изучения скоростей и механизмов биохимических процессов. Скорость реакции, средняя скорость реакции в интервале, истинная скорость. Факторы, влияющие на скорость реакции. Химическая кинетика изучает скорости химических реакций, их зависимость от различных факторов и механизмы реакций. Последовательность и характер стадий химических реакций называют механизмом реакции. Скорость химической реакции (v) определяется изменением концентрации Ас реагирующих веществ (или продуктов реакции) в единицу времени. Размерность скорости химической реакции — концентрация/время: Скорость химической реакции в общем случае не является постоянной в течение всего времени ее протекания. Рассмотрим обратимую реакцию: Истинная скорость (в любой момент времени) определяется первой производной концентрации по времени: Скорость химической реакции зависит в первую очередь от природы реагирующих веществ. Скорость гомогенной реакции зависит от концентрации реагентов, а гетерогенных — от площади соприкасающихся фаз, т. е. степени дисперсности. Скорости всех реакций зависят от температуры, многих реакций — от присутствия катализаторов. Классификации реакций, применяющиеся в кинетике: реакции, гомогенные, гетерогенные и микрогетерогенные; реакции простые и сложные (параллельные, последовательные, сопряженные, цепные). Молекулярность элементарного акта реакции. Кинетические уравнения. Порядок реакции. Период полупревращения. По фазовому состоянию реагентов реакции бывают гомогенные (однородные) и гетерогенные (неоднородные). В гомогенных реакциях все взаимодействующие вещества находятся в одной фазе (газовой, жидкой или твердой). Зоной реакции при проведении гомогенных реакций служит весь реакционный объем. В гетерогенных процессах реагенты, принимающие участие в реакции, находятся в разных фазах. В реакционном объеме одновременно находятся две или более фазы, а химическая реакция протекает на границе раздела фаз или в объеме одной из фаз.
По механизму различают простые и сложные реакции. Простые реакции осуществляются посредством однотипных элементарных актов. Под элементарным актом понимают единичный акт взаимодействия или превращения частиц, в результате которого образуются новые частицы продуктов реакции или промежуточных соединений. В элементарном акте принимает участие одна или две частицы (описаны единичные случаи одновременного взаимодействия трех частиц). Для осуществления сложных реакций необходимы разнотипные (не менее двух) элементарные акты. Различают следующие типы сложных реакций: параллельные, последовательные, сопряженные, цепные. Для параллельных реакций характерно протекание нескольких процессов с участием одних и тех же исходных веществ. Эти процессы завершаются образованием разных продуктов реакции. Скорость параллельных реакций определяется наиболее быстрой стадией. В последовательных реакциях образование конечного продукта реакции из исходных веществ происходит не непосредственно, а через ряд промежуточных продуктов. Скорость последовательной реакции определяется наиболее медленной стадией, которая называется лимитирующей. Некоторые сложные реакции состоят как из последовательных, так и параллельных В любом элементарном акте участвует одна, две или (очень ред- ко) три частицы. Таким образом, для каждой элементарной реакции можно указать её молекулярность. В мономолекулярных реакциях участвует одна молекула, кото- рая либо распадается на две части, либо перегруппировывает свои атомы. В бимолекулярном процессе происходит столкновение двух частиц, что приводит к их химическому превращению. Тримолекулярные элементарные реакции, в которых происхо- дит одновременное столкновение трёх частиц с последующим обра- зованием продуктов, обнаруживаются редко. Вероятность истинной тримолекулярной стадии в газовой фазе очень мала: тройные столк- новения происходят гораздо реже (1: 1000), чем двойные. реакций. Уравнение, описывающее зависимость скорости реакции (v) от концентрации (с)реагирующих веществ, называется кинетическим. Важной характеристикой реакции является период полупревращения t05 — время, за которое в реакцию вступает половина исходного вещества. Для радионуклидов аналогичная величина называется периодом полураспада.
13.Зависимость скорости реакции от концентрации. Кинетические уравнения реакций первого, второго и нулевого порядков. Экспериментальные методы определения скорости и константы скорости реакций. Скорость гомогенной хим. Реакции определяется изменением концентрации реагирующих веществ (или продуктов реакции) в единицу времени. Скорость прямой реакции уменьшается по мере расходования исходных веществ, а скорость обратной реакции увеличивается по мере накопления продуктов реакции. Когда скорости прямой и обратной реакции сравняются, система перейдет в состояние химического равновесия. Истинная скорость в любой момент времени является только положительной величиной и определяется первой производной концентрации по времени V= - Средняя скорость: Vср = Зависимость скорости хим.реакции от концентрации описывается кинетическим уравнением. Порядок реакции показывает, каким образом скорость реакции зависит от концентрации реагентов. Порядок кинетического уравнения может принимать значения 0, 1, 2 и 3; он может быть также дробным. Уравнения нулевого порядка: С0– с1=kt; C0-0.5C0=kt0.5 ; T0.5= Где с0– начальная концентрация реагента; с1 – концентрация реагента в момент времениt; k– константа скорости реакции; T0.5 – период полураспада. Уравнения первого порядка: ln= T0.5= Где ct=0.5c0 Уравнения второго порядка: T0.5= Закон действующий масс(Закон Гульберта и Вааге): скорость химической реакции пропорциональна произведению концентраций реагирующих веществ в степени их стехиометрических коэффициентов. Т.е. чем больше концентрация, тем больше скорость хим.реакции. Измерение скорости реакции основано на определении концентрации одного из реагирующих веществ через различные промежутки времени от начала реакции. Для определения концентраций можно применять методы физико-химического анализа, основанные на зависимости физических свойств смеси от её состава (например, определение показателя преломления, угла вращения плоскости поляризации, вязкости, электрической проводимости, объёма, плотности, изменения температур замерзания и кипения, интенсивности окраски и т. п.), и методы аналитической химии (например, титрование). Поскольку концентрации по ходу реакции непрерывно меняются, то необходимо или очень быстрое измерение концентрации (методы физико-химического анализа), или торможение реакции во взятой пробе (химический контроль). Торможение может быть достигнуто охлаждением, резким разбавлением, устранением катализатора или совместным действием всех указанных факторов. Если реакция, протекающая в газовой фазе, сопровождается изменением числа молекул, то её течение удобно контролировать по изменению давления смеси во времени. К сравнительно медленным реакциям со временем полупревращения порядка получаса и более можно применять спектроскопию, масс-спектрометрию и хроматографию. Для исследования скоростей очень быстрых реакций (с периодом полупревращения до 10-7и даже 10–9с) используются специально разработанные методы и особая аппаратура.
14. Зависимость скорости реакции от температуры. Правило Вант - Гоффа. Температурный коэффициент скорости реакции и его особенности для биохимических процессов. Правило Вант-Гоффа: при повышении температуры на 10 градусов скорость гомогенной хим.реакции увеличивается в 2-4 раза.
где V2 — скорость реакции при температуре Т2, V1— скорость реакции при температуре Т1, Из уравнения Вант-Гоффа температурный коэффициент вычисляется по формуле:
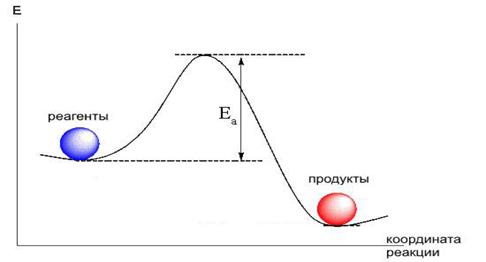
Теория активных соударений обобщает закономерности зависимости скорости хим.р-и от температуры: 1.Реагировать могут не все молекулы, а только находящиеся в особом активном состоянии 2.Активация молекулы происходит в результате биомолекулярного столкновения. 3.При столкновении частиц с примерно одинаковым запасом энергии происходит её перераспределение, в результате чего энергия одной из молекул достигает значения, соответствующего энергии активации. 4.Влияние температуры на скорость реакции: смещение равновесия между обычными и активными молекулами в сторону увеличения концентрации первых. Энергетический профиль реакции (график зависимости потенциальной энергии от координаты реакции)
Коллигативные свойства разбавленных растворов электролитов. Элементы теории растворов сильных электролитов Дебая- Хюккеля. Ионная сила, ее математическое выражение. Понятие об активности. Коэффициент активности. Активность ионов, а(Хi) – эффективная концентрация ионов (Хi), соответственно которой они проявляют себя в растворах сильных электролитов. Взаимосвязь между активностью ионова(Хi) и аналитической молярной концентрацией электролитас(Хi) выражается уравнением: а(Хi) = (Хi) где (Хi) -к оэффициент активности – величина, показывающая во сколько раз активность ионов отличается от их истинной аналитической концентрации (сi) в растворе сильного электролита. Величина (Хi) зависит от природы, температуры и концентрации электролитов в растворе. Для разбавленных растворов электролитов можно принять, что (Хi)=1, аа(Хi)=с(Хi), т.к. межионные взаимодействия практически отсутствуют и величина (Хi) в этом случае зависит от концентрации и заряда ионов, но практически не зависит от их природы. С увеличением концентрации величина (Хi) уменьшается, т.к. уплотняется ионная атмосфера. Для высоконцентрированных растворов (Хi)> > 1, т.к. гидратная оболочка ионов практически отсутствует и их подвижность возрастает. Рассчитать величину (Хi) можно по уравнению Дебая-Хюккеля: lgi =–Az i2 где А – коэффициент, зависящий от температуры; I-ионнаясила раствора; zi – заряд иона. Ионная сила раствора, I – величина, характеризующая интенсивность электростатического поля всех ионов в растворе, равная полусумме произведений молярной концентрации (сi) каждого иона на квадрат его заряда (zi)
Теория Дебая-Хюккеляприменима в диапазоне концентраций электролита0, 01-0, 05моль/л. Электролиты в организме..Осмоляльность и осмолярность биологических жидкостей и перфузионных растворов Понятия изо-, гипо-, гипертонический раствор. Понятие об изоосмии. Роль осмоса и осмотического давления в биологических системах. Плазмолиз. Цитолиз. Электролиты в организме: Na и Cl участвуют в поддержании кислотно-щелочного баланса, осмотического равновесия в организме. Са играет большую роль в построении костной ткани и зубов, в регулировании кислотности крови и ее свертывании, в возбудимости мышечной и нервной ткани. К находится преимущественно в жидкостях тела и мягких тканях, где является необходимым элементом для поддержания осмотического давления, регуляции рН крови. Mg является кофактором многих ферментативных реакций, необходим на всех этапах синтеза белка. В живых организмах Fe является важным микроэлементом, катализирующим процессы обмена кислородом. Сo входит в состав витамина В12, задействован при кроветворении, функциях нервной системы и печени, ферментативных реакциях. Zn необходим для метаболизмавитамина E, участвует в синтезе разных анаболических гормонов в организме, включая инсулин, тестостерон и гормон роста. Mn оказывает влияние на рост, образование крови и функции половых желёз. Плазма крови сложная многокомпонентная система, поэтому для учѐ та еѐ осмотических свойств было введено понятие осмолярной (осмолярность) или осмоляльной (осмоляльность) концентраций, разница между которыми незначительна вследствие относительной разбавленности биологических растворов. Осмолярная концентрация – количество всех кинетически активных частиц, содержащихся в 1 л раствора, независимо от их формы, размера и природы. сосм =i с(Х) G Осмолярная концентрация плазмы крови равна 0, 29–0, 31осмоль/л. Осмоляльность – концентрация осмотически активных частиц в растворе, выраженная в количестве осмоль на килограмм растворителя (осм/кг). Внорме осмоляльность плазмы (Опл) определяется концентрацией Na+, мочевины и глюкозы. Внорме осмоляльность плазмы составляет 275-290мосм/кг. Осмоляльность плазмы сохраняется постоянной благодаря механизмам, способным реагировать на изменения, равные1-2%ее исходной величины Осмоляльность является показателем осмотической концентрации и связана с числом растворенных частиц. Она определяется степенью диссоциации или, наоборот, ассоциации молекул, присутствующих в данной массе раствора. Осмоляльность выражается в (ммоль/кг.
Изотонические растворы – растворы с одинаковым осмотическим давлением. При контакте с изотоническим раствором осмотической ячейки, представляющей собой систему, отделѐ нную от окружающей среды мембраной с избирательной проницаемостью, между ними происходит равновесный обмен растворителем. Все клетки живых организмов являются осмотическими ячейками. Раствор, который изотоничен плазме крови, называется физиологическим, например, 0, 9%-ныйраствор NaCI. Гипертонический раствор – раствор, обладающий большим осмотическим давлением по сравнению с контактируемым раствором. При контакте осмотической ячейки с гипертоническим раствором наблюдается экзосмос - движение растворителя из осмотической ячейки в окружающий еѐ гипертонический раствор. Наблюдаемое при этом явление называется плазмолизом. Плазмолиз – сжатие и сморщивание клеток за счѐ т экзосмоса в гипертоническом растворе. При внутривенном введении больному гипертонического по отношению к плазме крови раствора происходит осмотический конфликт – обезвоживание и сморщивание клеток вследствие экзоосмоса. При резком плазмолизе клетки могут погибнуть. Гипертонические растворы используют для промывания гнойных ран, в качестве слабительных препаратов (горькая соль MgSO4∙ 7H2O, глауберова соль Na2SO4∙ 10H2O), диуретиков. Гипотонический раствор – раствор, обладающий меньшим осмотическим давлением по сравнению с контактируемым раствором. При контакте осмотической ячейки с гипотоническим раствором происходит эндосмос – движение растворителя в осмотическую ячейку из окружающего еѐ гипотонического раствора, приводящее к набуханию клетки и даже ее разрыву. Гемолиз – набухание и разрушение за счѐ т эндосмоса клеточных мембран эритроцитов в гипотоническом растворе, приводящее к выделе-
нию гемоглобина в плазму (лаковая кровь). Разрушение клеток называют лизисом. Изоосмия - относительное постоянство осмотического давления в жидких средах и тканях организма, обусловленное поддержанием на данном уровне концентраций содержащихся в них веществ: электролитов, белков и т. д. Это одна из важнейших физиологических констант организма, обеспечиваемых механизмами саморегуляции (гомеостаза) Цитолиз - разрушение животных и растительных клеток, выражающееся в полном или частичном их растворении.
Электронная теория Льюиса
Согласно теории Льюиса, кислотно-основные свойства соединений определяются их способностью принимать или отдавать пару электронов с образованием новой связи. Кислоты Льюиса – акцепторы пары электронов, основания Льюиса – доноры пары электронов. Кислотами Льюиса могут быть молекулы, атомы или катионы, обладающие вакантной орбиталью и способные принимать пару электронов с образованием ковалентной связи. К кислотам Льюиса относятся галогениды элементов II и III групп периодической системы, галогениды других металлов, имеющих вакантные орбитали, протон. Кислоты Льюиса в реакциях участвуют в качестве электрофильных реагентов. Основаниями Льюиса являются молекулы, атомы или анионы, имеющие неподеленную пару электронов, которую они предоставляют для образования связи с вакантной орбиталью. К основаниям Льюиса относятся спирты, простые эфиры, амины, тиоспирты, тиоэфиры, а также соединения, имеющие p-связи. В реакциях основания Льюиса проявляют себя как нуклеофильные частицы. Развитие теории Льюиса привело к созданию принципа жестких и мягких кислот и оснований (принцип ЖМКО или принцип Пирсона). Согласно принципа Пирсона, кислоты и основания подразделяются на жесткие и мягкие. Жесткие кислоты – это кислоты Льюиса, донорные атомы которых малы по размеру, обладают большим положительным зарядом, большой электроотрицательностью и низкой поляризуемостью. К ним относятся: протон, ионы металлов (К+, Na+, Mg2+, Ca2+, Al3+), AlCl3 и др. Мягкие кислоты - – это кислоты Льюиса, донорные атомы которых имеют большие размеры, большую поляризуемость, обладают малым положительным зарядом и низкой электроотрицательностью. К ним относятся: ионы металлов (Ag+, Cu+), галогены (Br2, I2), катионы Br+, I+ и др. Жесткие основания – основания Льюиса, донорные атомы которых обладают высокой электроотрицательностью, низкой поляризуемостью, имеют малый радиус атома. К ним относятся: Н2О, ОН-, F-, Cl-, NO3-, ROH, NH3, RCOO- и др. Мягкие основания - основания Льюиса, донорные атомы которых обладают высокой поляризуемостью, низкой электроотрицательностью, имеют большой радиус атома. К ним относятся: Н-, I-, C2H4, C6H6, RS- и др. Суть принципа ЖМКО состоит в том, что жесткие кислоты реагируют с жесткими основаниями, мягкие кислоты – с мягкими основаниями Ионизация слабых кислот и оснований. Константа кислотности и основности. Связь между константой кислотности и константой основности в сопряженной протолитической паре. Амфолиты. Изоэлектрическая точка.
Сила кислот определяется их способностью отдавать протон, а оснований — принимать его. Мерой этой способности служат соответственно константа кислотности Кa и константа основности Кb.
|
Последнее изменение этой страницы: 2017-03-15; Просмотров: 2452; Нарушение авторского права страницы

 , наиболее употребляемая единица измерения — моль на литр-секунду (моль/л*с)
, наиболее употребляемая единица измерения — моль на литр-секунду (моль/л*с)
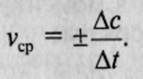
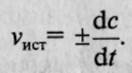
 . Если измерение истинной скорости проводят по изменению концентрации исходных веществ, то перед производной ставят знак минус:
. Если измерение истинной скорости проводят по изменению концентрации исходных веществ, то перед производной ставят знак минус: 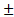
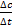
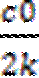 ;
;  = -kt;
= -kt; 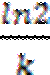 =
= 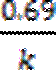 ;
; 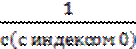 -
- 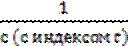 =kt;
=kt; 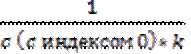 ;
; 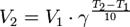
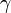 — температурный коэффициент реакции (если он равен 2, например, то скорость реакции будет увеличиваться в 2 раза при повышении температуры на 10 градусов).
— температурный коэффициент реакции (если он равен 2, например, то скорость реакции будет увеличиваться в 2 раза при повышении температуры на 10 градусов).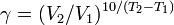
 с(Хi),
с(Хi), 
 I ,
I ,