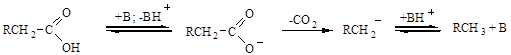|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Механизмы и кинетика кислотно-основного катализаСтр 1 из 5Следующая ⇒
Механизмы и кинетика кислотно-основного катализа Выполнила студентка группы ХТП-17-1м Павленина Виктория Проверил преподаватель: Баньковская Е.В.
Пермь 2018 ГОМОГЕННЫЙ КИСЛОТНО-ОСНОВНЫЙ КАТАЛИЗ
Множество реакций органической химии, в том числе и каталитических, может быть рассмотрена с позиции кислотно-основных взаимодействий. Трактовка механизмов кислотно-основного катализа (как гомогенного, так и гетерогенного) базируется на фундаментальных положениях теории кислот и оснований. Кислотами или основаниями считаются вещества, проявляющие кислотные или основные свойства. Проявление кислотных или основных свойств зависит от условий среды, в которой находятся эти вещества. Первый научный подход к определению кислот и оснований был сделан Оствальдом и Аррениусом в 1890 г. после создания последним теории электролитической диссоциации. Это была ионная теория, согласно которой кислота - это водородсодержащее соединение, способное генерировать протон (Н+), а основание - это вещество, генерирующее гидроксил (ОН-). МЕХАНИЗМЫ КИСЛОТНО-ОСНОВНОГО КАТАЛИЗА И ФАКТОРЫ, ОПРЕДЕЛЯЮЩИЕ ЕГО ЭФФЕКТИВНОСТЬ. Разделяют четыре типа гомогенных катализаторов действующих по механизмам кислотно-основного взаимодействия: 1) нуклеофильные; 2) кислотные; 3)основные; 4)электрофильные. НУКЛЕОФИЛЬНЫЙ КАТАЛИЗ Таблица 2.3. Нуклеофильность ЕNu в реакции нуклеофильного замещения при насыщенном атоме углерода и ее связь с основностью Н и поляризуемостью Р нуклеофила (Nu).
Из данных таблицы 2.3 видно, что такое сильное основание, как F- в данных условиях является очень слабым нуклеофилом из-за малой поляризуемости, а очень слабое основание I-, наоборот, проявляет высокую нуклеофильную активность. В тех случаях, когда основность является фактором, определяющим нуклеофильность (aP < < bH), первым членом в уравнении Эдвардса можно пренебречь и оно принимает вид уравнения Бренстеда: lg(kNu/kH2O) = bH = blg(КВNu/КВH2O) которое еще ранее было предложено Бренстедом в форме: kNu = G(КВNu)b или lg(kNu) = lg(G) + blg(КВNu) где lg(G) = lg(kH2O) - blg(КВH2O) Корреляционное уравнение Бренстеда хорошо применимо для реакций, контролируемых зарядами взаимодействующих атомов, а также в сериях реакций, в котором атом реакционного центра одинаков и его поляризуемость можно считать постоянной (Р = const), например для нуклеофильных реакций фенолятов, меркаптидов, аминов и др. В соответствии с уравнением (2.36) для серий таких реакций наблюдается линейная зависимость lg(kNu) от lg(КВNu) со значениями b в пределах 0< b< 1.
КИСЛОТНЫЙ КАТАЛИЗ В органическом синтезе наиболее распространен катализ протонными кислотами. Наиболее используемые из них: серная, соляная, орто-фосфорная, ароматические сульфокислоты (бензолсульфокислоты, толуолсульфокислота), муравьиная и некоторые другие. Очевидно, что кислотный катализатор способен взаимодействовать с реагентом в том случае, если последний проявляет основные свойства. Катализатор вступает с реагентом в кислотно-основное равновесие как с переносом заряда, так и без: Н + В ß à АН× × × ×: В ß à А-: × × × × НВ+ ß à А- + НВ+ Протонирование реагента приводит к появлению положительного заряда и сильной поляризации соседних связей, что так или иначе вызывает дефицит электронов на одном из атомов и делает возможным или облегчает дальнейшее превращение. Оно может протекать по двум механизмам: А-1 и А-2. Механизм А-1 включает стадию мономолекулярного распада протонированной молекулы по s-связи: RX + H+ ß à RXH+ ß à R+ + XH Стадия распада лимитирует скорость реакции. Такому механизму благоприятствуют электронодонорные заместители, стабилизирующие карбокатион, который затем по быстрой реакции взаимодействует либо с реагентом-нуклеофилом с образованием продукта замещения: R+ +: YH à RY + H+ либо с основанием, отщепляющим протон с образованием ненасыщенного соединения: Н-С-С+ + В à ВН+ + С=С Образующийся на первой стадии карбокатион иногда успевает изомеризоваться в более устойчивый. По механизму А-1 протекают реакции спиртов, простых и сложных эфиров, азотистых соединений, которые образуют стабильные промежуточные катионы. Например: Круг реакций, протекающих по механизму А-2 более широкий. Он включает активирование реагента не только за счет протонирования s-связанного заместителя (RXH+), но и за счет присоединения протона по кратным связям. Дальнейшее превращение протонированного соединения всегда бимолекулярно. Соединения, протонированные по s-связанному заместителю, подвергаются нуклеофильной атаке вторым реагентом по насыщенному атому углерода. Например: С2Н5-О-С2Н5 + Н3О+ ß à (С2Н5)2ОН+ + Н2О: ß à С2Н5ОН + С2Н5ОН2+ Для соединений, протонированных по кратным связям, характерны последующее присоединение нуклеофила и отщепление протона: +Н2О СН3СН=СН2 + Н+ ß à (СН3)2СН+ ß à (СН3)2СНОН2+ ß à (СН3)2СНОН + Н+ или последующее присоединение по ароматическим или кратным связям с последующим отщеплением протона:
В реакциях производных карбоновых кислот (RСОX) присоединение нуклеофильного реагента к протонированной карбоксильной группе приводит к последующему отщеплению группы Х и образованию продукта замещения. Например, как при гидролизе сложных эфиров:
Кислотный катализ без полной передачи протона от кислоты к реагенту характерен для слабых оснований-реагентов, слабых кислот-катализаторов и неполярных растворителей. В таких случаях кислотно-основное взаимодействие реагента с катализатором останавливается на стадии образования водородных связей. Например, в отсутствии добавок сильных кислот роль кислотного катализатора при этерификации уксусной кислоты спиртом выполняет вторая молекула уксусной кислоты:
Рисунок 2.3. Кинетический тест на наличие общего или специфического катализа: сплошные линии - общий катализ; пунктир - специфический катализ; рН1 > рН2. Общий кислотный катализ При общем кислотном катализе скорость реакции зависит от концентрации каждой из форм кислотного катализатора, присутствующей в реакционной массе. Общий кислотный катализ проявляется в тех случаях, когда стадия присоединения протона к реагенту является лимитирующей, а дальше идут быстрые реакции превращения активированных молекул. В общем виде механизм реакций можно представить следующей схемой: медленно: быстро: Образовавшийся на первой (лимитирующей) стадии активированный реагент (RH+) далее быстро превращается в продукт реакции (P) по мономолекулярной реакции или при участии второго реагента (Y). В соответствии с данной схемой скорость реакции лимитируется протолитической реакцией (2.56) и кинетическое уравнение имеет вид: r = k1[R][AH] При этом, если катализатор присутствует в разных формах (AiH), то каждой из них соответствует своя константа скорости (ki), определяемая активностью данной формы катализатора, и, в общем виде, уравнение (2.57) можно представить так:
Рассмотренная схема характерна для реакций, в которых на первой стадии происходит медленное образование С-Н связи. Примером служат кислотно-каталитические реакции олефинов, протекающие с образованием карбокатионов:
Но бывают и другие случаи. Известно множество примеров, когда лимитирующей оказывается стадия передачи протона между атомами катализатора и реагента. Это происходит, когда перенос протона сопровождается синхронным разрывом или образованием новой связи. Этому обычно предшествует быстрая стадия координации кислоты и основания за счет образования водородной связи. Общая схема для таких случаев выглядит так: Образование водородной связи (быстро):
Лимитирующая стадия: мономолекулярная бимолекулярная
Согласно приведенной схемы (2.60), скорость реакции будет определяться скоростью второй (лимитирующей) стадии, и кинетическое уравнение (например, для бимолекулярного превращения) будет иметь следующий вид: r = k1[R× × × HA][Y] Но при высоких значениях константы равновесия первой стадии или при высоких концентрациях реагента, что обеспечивает количественное связывание катализатора в активированный комплекс с реагентом (т.е. когда [R× × × HA] = [HA]), уравнение принимает более простой вид: r = k1[HA][Y] Примерами бимолекулярных превращений активированного комплекса при общем кислотном катализе могут служить реакции этерификации:
ЭЛЕКТРОФИЛЬНЫЙ КАТАЛИЗ Электрофильный катализ осуществляется кислотами Льюиса. Электрофильными катализаторами в органических реакциях могут выступать нейтральные молекулы, соли металлов, ионы и некоторые другие соединения, например: Соли - ZnCl2, AlCl3, FeCl3, TiCl4, SnCl4, BF3; Ионы металлов - Li+, Ag+, Hg+; Молекулы и ионы - SO3, P2O5, R+, NO2+ Роль электрофильного катализатора заключается в активации реагента и аналогична роли протона в кислотном катализе. Взаимодействие электрофильного катализатора с реагентом-основанием заключается в образовании донорно-акцепторной связи за счет пары электронов реагента-основания, занимающей вакантную орбиталь одного из атомов катализатора. Электрофильные катализаторы оказываются гораздо более эффективными, чем протонные кислоты при активировании таких слабоосновных реагентов, как галогены, алкилгалогениды, ангидриды и галогенангидриды, эпоксиды и т.п. Образование донорно-акцепторного комплекса либо сильно поляризует связь в молекуле реагента, либо приводит к полному ее разрыву с образованием катиона:
Образовавшийся катион или поляризованная молекула легко вступает в дальнейшее химическое превращение:
В некоторых случаях кислота Льюиса выполняет каталитические функции, активируя не сам реагент, а протонную кислоту, являющуюся сокатализатором: HF + SbF5 à HSbF6 HF + BF3 à HBF4
Такие комплексы оказываются значительно более эффективными донорами протона (увеличение кислотности), чем исходные кислоты и относятся к разряду суперкислот (см. раздел Суреркислоты). В частности, комплекс HF× TaF5 обладает настолько высокой кислотностью, что отщепляет гидрид-анион от углеводородов с образованием карбокатиона: RCH2-CH3 + HF× TaF5 ß à RCH-CH3 + H2 + TaF6- Галогениды металлов (AlCl3, FeCl3, TiCl4, SnCl4, BF3) разлагаются при взаимодействии с кислород- или азотсодержащими органическими соединениями и водой либо образуют с ними очень прочные и малореакционноспособные комплексы. Поэтому галогениды металлов обычно не катализируют реакции с их участием. ОСНОВНЫЙ КАТАЛИЗ Основный катализ осуществляется веществами со свободной или лабильной электронной парой электронов, которые могут быть как анионами, так и нейтральными молекулами (HO-, RO-, NH2-, R3N: и др.). При этом реагент должен обладать кислотными свойствами (по отношению к катализатору), и активирование реагента осуществляется за счет отрыва от него протона или образования водородной связи:
Причем в активации реагента могут принимать участия все формы оснований, образующиеся в реакционной массе, в том числе и ионы лиата, образующиеся по протолитической реакции катализатора с растворителем: SH + B- ß à S- + BH После активации реагента (уравнение (2.69)) превращения могут протекать по двум вариантам: 1) Мономолекулярная реакция. Из продуктов превращений на последующих стадиях регенерируется катализатор. Например:
2)Бимолекулярная реакция. Участие активированного реагента в качестве нуклеофила в реакции со вторым реагентом. Эти реакции близки по механизму к нуклеофильному катализу (схема (2.27)). Отличие состоит в том, что при основном катализе протекает еще одна стадия в каталитическом цикле - отщепление катализатором протона от реагента с образованием сопряженного основания реагента, играющего далее роль нуклеофильного катализатора, но с обязательной регенерацией основного катализатора в конце каталитического цикла. Например в реакции альдольной конденсации:
В других случаях не происходит полного отщепления протона, а нуклеофильность реагента повышается за счет образования водородной связи с основанием-катализатором:
Механизмы и кинетика кислотно-основного катализа Выполнила студентка группы ХТП-17-1м Павленина Виктория Проверил преподаватель: Баньковская Е.В.
Пермь 2018 |
Последнее изменение этой страницы: 2019-05-06; Просмотров: 1049; Нарушение авторского права страницы



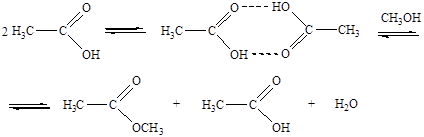
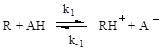



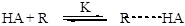

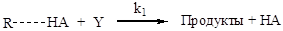

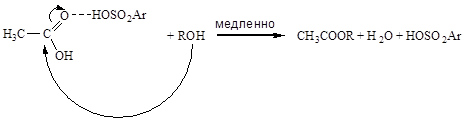




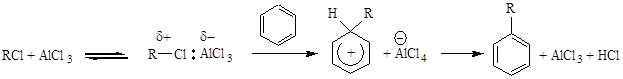
 или
или