
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
КОНЦЕПЦИИ ПРОЦЕССОВ И ВОЗМОЖНОСТИ УПРАВЛЕНИЯ ИМИ
8.1. Химический катализ и методы управления химическими процессами Реакционная способность вещества на 50 % определяется его составом и структурой и на 50 % — его реагентом по реакции. Так, если реагент — сильная кислота, то вещество ведет себя как основание, и наоборот. Эту двойственность поведения в реакции объяснил Бутлеров, считая, что вещество расщепляется на два изомера и это влияет на равновесную изомерию (таутомерия). Впоследствии А. Н. Несмеянов установил, что это — раздвоение свойств изомера как целого. К 70-м годам XIX в. идеи и методы физики стали проникать в смежные области естествознания. Н. Н. Бекетов впервые сформулировал и обосновал положение о том, что физическая химия — самостоятельная наука, основная задача которой состоит в изучении связи между физическими и химическими свойствами веществ, явлений и процессов. Работами М.Бертло, Ю.Томсона, В.Ф.Лучинина и Н.Н.Бекетова была создана термохимия, изучающая закономерности в теплоте образования и сгорания веществ в зависимости от их химического строения. Исследования Дж. Гиб-бса, Я. Г. Вант-Гоффа, В.Г.Нернста и других ученых развили химическую термодинамику, изучающую энергетические процессы, которые сопровождают процессы химические. Гиббс сформулировал правило фаз, по которому система имеет несколько состояний, разделенных между собой границами. Нернст установил, что при приближении к температуре, равной 0 К, тепловой эффект и движущая сила химических реакций все более совпадают и это дает возможность производить точный расчет химических реакций. Начало систематическому расчету реакций положил Н.А. Меншуткин. Химические реакции — основа химии. При столкновениях молекул может высвободиться энергия, достаточная для перегруппировки электронов в них и формирования нового набора связей, т. е. образования новых соединений. Химические реакции обычно представляют в виде уравнений: слева — исходные вещества, справа — продукты реакции; стрелка обращена в сторону более низкой суммарной энергии связей, показывая, в какую сторону реакция стремится идти самопроизвольно. Но реакции могут идти в обе стороны и представляют собой перераспределение химических связей. Исследования общих закономерностей, управляющих химическими процессами, заинтересовали возникшую в конце XIX в. химическую индустрию. Если какое-то вещество является катализатором, или ингибитором, происходит целый комплекс реакций, участвуют все вещества, оказавшиеся в зоне реакции, и могут получиться различные побочные продукты. От знания скорости и направления реакций, влияния на них различных факторов зависела производительность химической промышленности. Определение характера химического процесса казалось почти невозможным, пока не создали химическую термодинамику и кинетику. Ответ на вопрос, от чего зависит возможность осуществления реакции, перестройки химических связей дают законы термодинамики. Например, для получения теплоты требуется определенное топливо. Переход теплоты от нагретого тела связан с распределением энергии: атомы вещества отдают энергию теплового движения окружающим атомам, не меняя своего состояния. При химических реакциях энергия тоже рассеивается, но меняются окружение атомов и исходное вещество, может возникнуть новое вещество. При решении разнообразных термодинамических задач используют особые функции — термодинамические потенциалы. Зная выражение термодинамических потенциалов, через независимые параметры системы можно вычислить и другие характеристики процессов. Приведем некоторые из них. Подставив в выражение для первого начала термодинамики в обратимом процессе
Это выражение, объединяющее первое и второе начала термодинамики, является полным дифференциалом внутренней энергии, а общее уравнение для полного дифференциала таково: Итак, частная производная от внутренней энергии по энтропии равна температуре, взятая с обратным знаком производная по объему равна давлению, а сама внутренняя энергия является термодинамическим потенциалом. Другой термодинамический потенциал ввел Г.Гельмгольц (1877), он показал, что функция
Найдем полный дифференциал свободной энергии:
лучаем
Физический смысл свободной энергии F ясен из выражения для dF. При ние свободной энергии равно работе, совершаемой системой в изотермическом процессе. Сохранение постоянной температуры тела у живых организмов позволяет считать, что производимая ими работа совершается за счет уменьшения свободной энергии. Важным для химических процессов является и термодинамический потенциал, так называемая функция Гиббса — + Vdp. Сравнивая полученное уравнение с выражением для полного дифференциала, запишем:
Потенциал Гиббса используют при расчетах энтропии и объема в изобарно-изотермических процессах. При стремлении системы к равновесию в необратимом изобарно-изотермическом процессе
При изменении числа частиц в системе вводят так называемый химический потенциал
Итак, термодинамический потенциал равен изменению потенциала, приходящегося на одну частицу в соответствующем процессе. И реакция возможна, если она сопровождается уменьшением величины потенциала. Когда камень падает в поле тяготения, уменьшается его потенциальная энергия. Подобный процесс наблюдается и в химической реакции: когда она идет, ее свободная энергия переходит на более низкий уровень. В этих примерах аналогия полная, поскольку нет изменения энтропии. Но в химических реакциях изменение энтропии необходимо учитывать, и возможность реакции еще не означает, что она пойдет самопроизвольно. Термодинамика объясняет: реакция пойдет только при уменьшении энергии веществ и увеличении энтропии. Энтропия растет, так как в малой молекуле расположение атомов менее упорядочено, чем в большой. Но реальные процессы и состояния чаще всего являются неравновесными, а системы — открытыми. Такие процессы рассматриваются в неравновесной термодинамике. В 1886 г. появилась работа Гиббса, содержащая правило фаз и новые методы исследования и описания условий равновесия через термодинамические потенциалы. При термодинамическом подходе управление ходом реакции осуществляется изменением термодинамических параметров системы — температуры, давления, концентрации. Этими параметрами можно сместить направление реакции. Но термодинамический подход не дает изменения скорости реакции, так как время не входит в уравнения. Поэтому сведения о скорости дает только кинетика. Закон действующих масс открыли и представили в математической форме норвежские ученые К. М. Гульберг и П. Вааге (1879). Они, рассматривая равновесие как динамический процесс, построили теорию скоростей химических реакций. Вант-Гофф ввел в теорию принятый сейчас термин «концентрация». Если реакция происходит между компонентами А, В, С, этот закон принимает следующий вид: скорость реакции станта скорости реакции (в скобках — концентрации реагентов), а, р, у — коэффициенты степени участия. Изменяя концентрации, можно менять скорость и направление реакции, т.е. управлять процессом. Выразив константу равновесия через концентрации реагентов, Вант-Гофф подвел теоретический фундамент под закон действующих масс и обосновал важнейшие положения химической кинетики (1884). Ле Шателье выдвинул принцип подвижного равновесия (1884). Сейчас его формулируют так: внешнее воздействие, которое выводит систему из состояния термодинамического рав- I новесия, вызывает в ней процессы, направленные на ослабление результатов такого влияния. Появилась возможность смещать равновесие в сторону образования продуктов реакции через изменение температуры, давления и концентрации реагентов. Эти методы назвали термодинамическими. Среди соединений реагентов есть образования с разной степенью устойчивости. Менее устойчивые обладают большей свободной энергией, значит, вновь образованная группировка менее устойчива, чем исходные компоненты. Для преодоления этой разницы в значениях свободной энергии нужна дополнительная энергия, так называемая энергия активации. Она определяет скорость реакции, и если ее недостаточно для преодоления барьера, реакция не идет. Снижают энергию активации катализаторами. Явление химического катализа было открыто в 1812 г. Кирхгофом. В XVIII в. уже знали о каталитическом действии селитры при получении серной кислоты, хотя смысл этого явления не поддавался объяснению. Берцеллиус связал природу взаимодействия агентов с электрохимическими потенциалами (1835), обозначив силу «вызывания химической деятельности» понятием каталитической силы. Либих предположил, что взаимодействие с катализатором может непрерывно менять химические связи в молекуле. Взгляды Либиха поддержал Д. И. Менделеев. К концу XIX в. поняли, что в реакции участвуют стенки сосуда, растворители и случайные примеси. Целенаправленное изучение катализа позволило к середине XX в. получать широкий круг органических продуктов, регулировать скорость и заданную направленность химических реакций. Д.П.Коновалов положил начало физико-химической теории катализа, ввел понятие активной поверхности (1885) и вывел формулу для скорости автокаталитических реакций независимо от С. Оствальда. Теорией катализа занимался и Д. И. Менделеев (1886). При катализе происходит активация молекул реагента при контакте с катализатором: связи в веществе становятся более подвижными, «подталкивая» вещества к взаимодействию. В. Оствальд, сравнивая относительную активность различных кислот, пришел к выяснению условий химического равновесия и развитию катализа. Он определил катализатор как вещество, «которое изменяет скорость реакции, но не входит в состав конечного продукта реакции». Доля каталитических процессов в химической промышленности достигает 80 %. За 50 лет катализ превратился в мощное орудие синтеза веществ. Зависимость скорости реакций от температуры исследовал С.Аррениус, предложивший (1889) закон: вероятность накопления энергии активации определяется формулой, полученной Больцманом: пии веществ, можно определить условия протекания реакции и ее направление. Природный катализатор — хлорофилл — комплексное металло-органическое соединение в живой ткани зеленого листа. Поэтому можно считать, что процесс фотосинтеза происходит при фото-биокатализаторе, и эти реакции изучаются в целях получения еще одного источника энергии. За идеями строения эффективных биокатализаторов химики часто обращаются к живой природе. Поэтому будущее катализа — на пути между химией и биологией. Большинство биохимических процессов — каталитические. Расчет энергии активации проводится в квантовой химии. Биокатализаторы были открыты в начале XX в. Благодаря работам французских химиков П.Сабатье и Ж.Б.Сандерана в промышленности при гидрировании органических веществ вместо благородных металлов стали использовать никель, медь, кобальт, железо. Русский химик-органик В.Н.Ипатьев исследовал каталитическое действие оксидов металлов при высоких давлениях и температурах и установил, что при использовании смеси катализаторов их действие усиливается. Каталитический способ синтеза аммиака из атмосферного азота и водорода под давлением открыл немецкий химик Ф. Габер. Затем химик-технолог К. Бош и А. Митташ предложили промышленный способ синтеза аммиака с использованием смеси катализаторов — железа, едкого калия и глинозема — при повышенных температурах и высоком давлении. Подбор катализаторов долгое время шел эмпирическим путем, да и сами механизмы химических реакций, особенно быстрых, не поддавались объяснениям. Наиболее быстро происходят реакции фотохимического типа, когда фотоны, разрушая равновесие, вызывают новые реакции и создают новые условия равновесия, что устанавливали по вторичному рассеянию молекул. В 20-е гг. такие реакции начал исследовать английский физико-химик Р.Дж.Норриш, но успех пришел к нему только после 1945 г., когда он привлек к работе молодого сотрудника Дж. Портера, специалиста по электронике и радиолокации. Они использовали сверхкороткие световые импульсы мощностью 600 МВт, излучаемые за 10-6 с. Впоследствии ученые уменьшили временной интервал до 10-9 с и стали использовать лазеры, имеющие мощность, в тысячи раз большую. Квантовая химия позволила рассмотреть на микроуровне протекание реакций, отдельные молекулы и их электронные структуры. Термодинамический подход, описывающий систему в целом, позволяет глубже понять тенденции течения реакций. В 1952 г. японский физико-химик Кэнити Фукуи продолжил работу над методом молекулярных орбиталей и пришел к оценке важности переходного состояния, или активированного комплекса, подчеркнув особую роль внешних электронов в течении реакций, в том числе и каталитических. Расчеты на ЭВМ подтвердили его соображения, идеи Фукуи используются и сейчас. Американский физико-химик Роалд Хофман, доведя метод Фукуи до расчетного, разработал правила сохранения орбитальной симметрии молекул при химических реакциях. Управлять ходом химической реакции можно и за счет привлечения внешнего источника энергии — световой или тепловой. С ее помощью удается расшатать атомы в исходной молекуле и побудить их к участию в нужной реакции. Этим занимается область химии, получившая название химии экстремальных состояний. Использованием для этой цели более жесткого электромагнитного излучения (для молекул с крепкими внутримолекулярными связями) занимается радиационная химия. Популярное:
|
Последнее изменение этой страницы: 2017-03-09; Просмотров: 460; Нарушение авторского права страницы
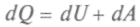 формулы для работы
формулы для работы 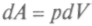 и количества теплоты
и количества теплоты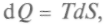 получим
получим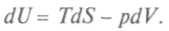 (1)
(1)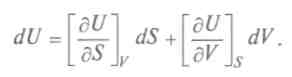
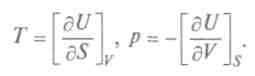
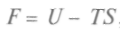 , называемая свободной энергией, может быть критерием термодинамического равновесия.
, называемая свободной энергией, может быть критерием термодинамического равновесия.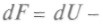
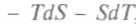 , и, используя выражение (1), можно записать:
, и, используя выражение (1), можно записать: 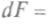
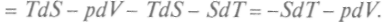 Учитывая (как и ранее), что dF является полным дифференциалом от переменных
Учитывая (как и ранее), что dF является полным дифференциалом от переменных 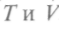 , по-
, по-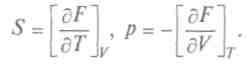
 , т.е. уменьше-
, т.е. уменьше-
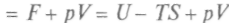 . Продифференцировав, получим:
. Продифференцировав, получим: 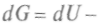
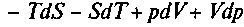 . С учетом (1) последнее уравнение можно переписать так:
. С учетом (1) последнее уравнение можно переписать так: 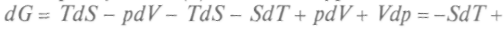
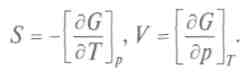
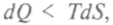 и для дифференциала Гиббса используют уже вместо написанного выше равенства следующее:
и для дифференциала Гиббса используют уже вместо написанного выше равенства следующее: 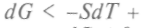
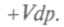 Но поскольку в этом процессе
Но поскольку в этом процессе 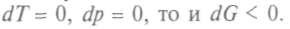 И это будет выполняться до установления равновесного состояния, когда и dG станет равно нулю. Можно сказать, что в неравновесных изобарно-изотермических процессах функция Гиббса убывает до минимума в состоянии равновесия. В изотермических процессах, происходящих без изменения объема, убывает также потенциал Гельмгольца — свободная энергия.
И это будет выполняться до установления равновесного состояния, когда и dG станет равно нулю. Можно сказать, что в неравновесных изобарно-изотермических процессах функция Гиббса убывает до минимума в состоянии равновесия. В изотермических процессах, происходящих без изменения объема, убывает также потенциал Гельмгольца — свободная энергия.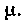 Тогда вместо уравнения (1) следует писать:
Тогда вместо уравнения (1) следует писать: 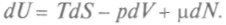 Здесь dN — изменение числа частиц в системе. Соответственно изменятся и выражения для других потенциалов:
Здесь dN — изменение числа частиц в системе. Соответственно изменятся и выражения для других потенциалов: 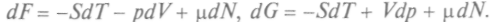 Тогда для химического потенциала при постоянных парах соответствующих параметров
Тогда для химического потенциала при постоянных парах соответствующих параметров 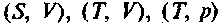 можно записать:
можно записать: 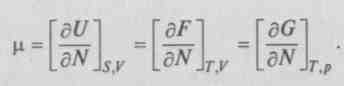
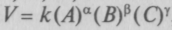 , где k — кон-
, где k — кон-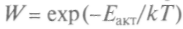 . Вант-Гофф исследовал причины, меняющие скорость реакций, и показал, что с ростом температуры энергия частиц при столкновениях может оказаться достаточной для начала химической реакции. Зная величины энтро-
. Вант-Гофф исследовал причины, меняющие скорость реакций, и показал, что с ростом температуры энергия частиц при столкновениях может оказаться достаточной для начала химической реакции. Зная величины энтро-