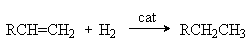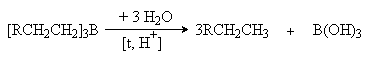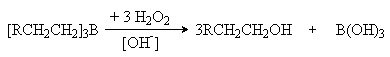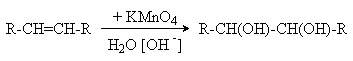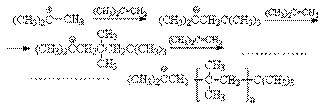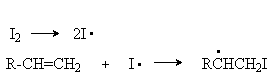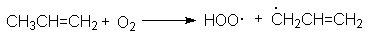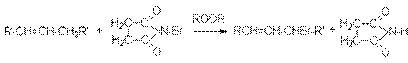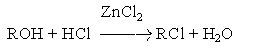|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Лекция 1 на тему: Предмет органической химии, ее значение. Теория химического строения органических соединений. Классификация и номенклатура органических соединений.Стр 1 из 17Следующая ⇒
Лекция 1 на тему: Предмет органической химии, ее значение. Теория химического строения органических соединений. Классификация и номенклатура органических соединений. Органическая химия изучает соединения углерода, называемые органическими веществами . В связи с этим органическую химию называют также химией соединений углерода. Органическая химия может быть определена также как химия углеводородов и их производных. Хотя такое определение более четко отражает содержание предмета органической химии, однако и оно не дает возможности провести резкую границу между органическими и неорганическими веществами. Неудачи многочисленных попыток найти эту границу привела многих химиков к мысли, что хотя неорганические и органические соединения следует рассматривать как строго самостоятельные группы, различие между ними довольно трудно определить. Эти неудачи объясняются стремлением дать формально-логическое определение органической химии, отыскать резкую грань между ней и неорганической химией. Такая задача ошибочна в самой ее постановке и принципиально неразрешима. Так как в природе все явления взаимосвязаны, то естественно, что грубое отсечение одной отрасли науки от другой невозможно. Между смежными науками существуют естественные диалектические переходы. В частности, на границе между органическими и неорганическими соединениями находятся такие вещества, как сода, сероуглерод, мочевина, оксид углерода (IV) и т. д., которые можно с равным правом рассматривать в качестве как органических, так и неорганических соединений. Место органической химии в ряду других наук определяется не только ее соседством с неорганической химией. Изучая сложнейшие органические вещества, играющие важную роль в жизнедеятельности животных и растительных организмов, органическая химия тесно соприкасается и с биологией. В пограничной между этими двумя науками области возникла и успешно развивается новая молодая наука - биологическая химия. Наконец, вследствие все расширяющегося в настоящее время применения физических методов исследования органических веществ теснее становится связь органической химии с физикой. Важнейшие причины выделения органической химии в отдельную науку заключаются в следующем: · Число известных органических соединений (около 5 миллионов) значительно превышает число соединений всех остальных элементов периодической системы Менделеева. В настоящее время известно около 650 тысяч неорганических соединений, примерно 200 тысяч новых органических соединений получают сейчас в один год. Это объясняется не только тем, что химики особенно интенсивно занимаются синтезом и исследованием органических соединений, но и особой способностью элемента углерода давать соединения, содержащие практически неограниченное число атомов углерода, связанных в цепи и циклы. · Органические вещества имеют исключительное значение вследствие их крайне многообразного практического применения, а особенно потому, что они играют важную роль в процессах жизнедеятельности организмов. · Имеются существенные отличия в свойствах и реакционной способности органических соединений от неорганических, вследствие чего возникла необходимость в развитии многих специфических методов исследования органических соединений.
История развития органической химии Органическая химия как наука оформилась в начале XIX в., однако знакомство человека с органическими веществами и применение их для практических целей началось еще в глубокой древности. 1. Первой известной кислотой был уксус, или водный раствор уксусной кислоты. 2. Древним народам было известно брожение виноградного сока, 3. Они знали примитивный способ перегонки и применяли, его для получения скипидара; галлы и германцы знали способы варки мыла; 4. В Египте, Галлии и Германии умели варить пиво. В Индии, Финикии и Египте было весьма развито искусство крашения при помощи органических веществ. 5. Кроме того, древние народы пользовались такими органическими веществами, как масла, жиры, сахар, крахмал, камедь, смолы, индиго и т. д. Период развития химических знаний в средние века (приблизительно до XVI в.) получил название периода алхимии . Однако изучение неорганических веществ было значительно более успешным, чем изучение веществ органических. Сведения о последних остались почти столь же ограниченными, как и в более древние века. Некоторый шаг вперед был сделан благодаря совершенствованию методов перегонки. Таким путем, в частности, было выделено несколько эфирных масел и получен крепкий винный спирт, считавшийся одним из веществ, с помощью которых можно приготовить философский камень. Конец XVIII в ознаменовался заметными успехами в изучении органических веществ, причем органические вещества начали исследовать с чисто научной точки зрения. В этот период был выделен из растений и описан ряд важнейших органических кислот (щавелевая, лимонная, яблочная, галловая), установлено, что масла и жиры содержат в качестве общей составной части «сладкое начало» - масел (глицерин) и т. д. Постепенно начали развиваться исследования органических веществ - продуктов жизнедеятельности животных организмов. Так, например, из мочи человека были выделены мочевина и мочевая кислота, а из мочи коровы и лошади - гиппуровая кислота. Накопление значительного фактическое материала явилось сильным толчком к более глубокому изучению органического вещества. Впервые понятия об органических веществах и об органической химии ввел шведский ученый Берцелиус (1827). Органическую химию он и определял как «химию растительных и животных веществ, или веществ, образующихся под влиянием жизненной силы». В 1828г Вёлер показал, что неорганическое вещество - циановокислый аммоний при нагревании превращается в продукт жизнедеятельности животного организма - мочевину. В 1845г Кольбе синтезировал типичное органическое вещество - уксусную кислоту, использовав в качестве исходных веществ древесный уголь, серу, хлор и воду. За сравнительно короткий период был синтезирован ряд других органических кислот, которые до этого выделялись только из растений, В 1854г Бертло удалось синтезировать вещества, относящиеся к классу жиров. В 1861г. А. М. Бутлеров действием известковой воды на параформальдегид впервые осуществил синтез метиленитана - вещества, относящегося к классу сахаров, которые, как известно, играют важную роль в процессах жизнедеятельности организмов. Теории органической химии Теория радикалов. Первая теория в органической химии - теория радикалов была тесно связана с электрохимической теорией Берцелиуса. Основываясь главным образом на факте электролиза неорганических соединений, Берцелиус считал, что все химические вещества состоят из электроотрицательных и электроположительных атомов или групп атомов, удерживаемых в молекуле силами электростатического притяжения. Фактическим основанием для создания теории радикалов послужили исследования соединений циана (Гей-Люссак, 1815). Этими исследованиями было впервые установлено, что при целом ряде химических превращений группа из нескольких атомов в неизменном виде переходит из молекулы одного вещества в молекулу другого, подобно тому, как переходят из молекулы в молекулу атомы элементов. Такие «неизменяемые» группы атомов получили название радикалов. На определенном этапе развития органической химии теория радикалов сыграла значительную роль, впервые дав химикам руководящую нить в исследовании органического вещества. Это оказалось возможным потому, что в основе теории радикалов лежало важное обобщение: при химических реакциях группы, атомов (т. е. радикалы) в неизменном виде входят в образующиеся при этих реакциях молекулы. Перегруппировка. Иногда сложная суммарная реакция может включать несколько типовых реакций, однако отдельные стадии суммарной реакции всегда можно отнести к одному из перечисленных типов. Как правило, основное органическое вещество, участвующее реакции, называют «субстратом», тогда другой компонент реакции обычно условно рассматривают как «реагент». Распределение электронной плотности в реагирующей молекуле часто определяет тип реагента, с которым основное органическое вещество (субстрат) будет реагировать. Так, в бромистом этиле углеродный атом, связанный с атомом брома и имеющий низкую электронную плотность, будет легко подвергаться атаке частицами, несущими отрицательный заряд (NС-, НО-), или молекулами, в которых имеются центры с высокой электронной плотностью (: NН3, : NR3):
δ + δ - СН3 - СН2→ Вr + ОН- → СН3 - СН2 - ОН + Вr- Субстрат нуклеофил В этом случае реагент называют нуклеофильным реагентом или нуклеофилом, а реакция называется нуклеофильной реакцией . Напротив, реагент с электронным дефицитом (чистый катион Н+, +NО2, С6Н5N2+) или молекула, имеющая центр с низкой электронной плотностью - SО3 и ВF3) будет реагировать с субстратом, в котором имеются центры с высокой электронной плотностью/ Реагент такого типа называется электрофильным реагентом или электрофилом, а реакция называется электрофильной реакцией . И электрофильные, и нуклеофильные реакции называются гетеролитическими реакциями . Существуют реакции, в которых реагент является радикальной частицей, несущей неспаренный электрон. Такие реакции называются радикальными или гомолитическими реакциями . Изомерия. Номенклатура. Алканы с n=1, 2, 3 могут существовать только в виде одного изомера
Начиная с n=4, появляется явление структурной изомерии.
Число структурных изомеров алканов быстро растет с увеличением числа углеродных атомов, например, пентан имеет 3 изомера, гептан - 9 и т.д. Число изомеров алканов увеличивается и за счет возможных стереоизомеров. Начиная с C7Н16 возможно существование хиральных молекул, которые образуют два энантиомера. Номенклатура алканов. Доминирующей номенклатурой является номенклатура IUPAC. В тоже время в ней присутствуют элементы тривиальных названий. Так, первые четыре члена гомологического ряда алканов имеют тривиальные названия. СН4 - метан С2Н6 - этан С3Н8 - пропан С4Н10 - бутан. Названия остальных гомологов образованы от греческих латинских числительных. Так, для следующих членов ряда нормального (неразветвленного) строения используются названия: С5Н12 - пентан, С6Н14 - гексан, С7Н18 - гептан, С8Н18 - октан, С9Н20 - нонан, С10Н22 - декан, С11Н24 - ундекан, С12Н26 - додекан, С13Н28 - тридекан, С14Н30 - тетрадекан, С15Н32 - пентадекан и т.д. Основные правила IUPAC для разветвленных алканов а) выбирают наиболее длинную неразветвленную цепь, название которой составляет основу (корень). К этой основе прибавляют суффикс “ан” б) нумеруют эту цепь по принципу наименьших локантов, в) заместитель указывают в виде префиксов в алфавитном порядке с указанием места нахождения. Если при родоначальной структуре находятся несколько одинаковых заместителей, то их количество указывают греческими числительными. В зависимости от числа других углеродных атомов, с которыми непосредственно связан рассматриваемый углеродный атом, различают: первичные, вторичные, третичные и четвертичные углеродные атомы.
Пример:
В качестве заместителей в разветвленных алканах фигурируют алкильные группы или алкильные радикалы, которые рассматриваются как результат отщепления от молекулы алкана одного водородного атома. Название алкильных групп образуют от названия соответствующих алканов путем замены последних суффикса “ан” на суффикс “ил”. СН3 - метил СН3СН2 - этил СН3СН2СН2 - пропил Для названия разветвленных алкильных групп используют также нумерацию цепи:
Начиная с этана, алканы способны образовывать конформеры, которым соответствует заторможенная конформация. Возможность перехода одной заторможенной конформации в другую через заслоненную определяется барьером вращения. Определение структуры, состава конформеров и барьеров вращения являются задачами конформационного анализа. 1. Фракционная перегонка природного газа или бензиновой фракции нефти. Таким способом можно выделять индивидуальные алканы до 11 углеродных атомов. 2. Гидрирование угля. Процесс проводят в присутствии катализаторов (оксиды и сульфиды молибдена, вольфрама, никеля) при 450-470оС и давлениях до 30 Мпа. Уголь и катализатор растирают в порошок и в суспензированном виде гидрируют, борботируя водород через суспензию. Получающиеся смеси алканов и циклоалканов используют в качестве моторного топлива. 3. Гидрирование СО и СО2. СО + Н2 алканы СО2 + Н2 алканы В качестве катализаторов этих реакций используют Со, Fe, и др. d - элементы. 4. Гидрирование алкенов и алкинов.
В качестве катализаторов используют Ni, Pt, Pd. 5. Металлоорганический синтез. а). Синтез Вюрца. 2RHal + 2Na R R + 2NaHal Этот синтез малопригоден, если в качестве органических реагентов используют два разных галогеналкана. б). Протолиз реактивов Гриньяра. R Hal + Mg RMgHal RMgHal + HOH RH + Mg(OH)Hal в). Взаимодействие диалкилкупратов лития (LiR2Cu) с алкилгалогенидами LiR2Cu + R X R R + RCu + LiX Сами диалкилкупраты лития получают двухстадийным способом
2R Li + CuI LiR2Cu + LiI 6. Электролиз солей карбоновых кислот (синтез Кольбе). 2RCOONa + 2H2O R R + 2CO2 + 2NaOH + H2
7. Сплавление солей карбоновых кислот со щелочами.
Реакция используется для синтеза низших алканов. 8. Гидрогенолиз карбонильных соединений и галогеналканов. а). Карбонильные соединения. Синтез Клемменса.
б). Галогеналканы. Каталитический гидрогенолиз.
В качестве катализаторов используют Ni, Pt, Pd. в) Галогеналканы. Реагентное восстановление. RHal + 2HI RH + HHal + I2 Физические свойства алканов Алканы представляют собой бесцветные вещества, в обычных условиях газообразные или жидкие. Алканы с большим числом углеродных атомов (nC > 15) – твердые вещества. Жидкие и твердые алканы намного легче воды. Молекулы алканов представляют собой совокупность -связей (С-Н и С-С), характеризующихся высокой прочностью, низкими полярностью и поляризуемостью. Возбуждение молекул алканов требует большой энергии, поэтому они поглощают только в вакуум-УФ-области (125-140 нм) и являются хорошими растворителями для снятия электронных спектров различных веществ. В колебательных спектрах (ИК-спектры и спектры комбинационного рассеяния) для алканов характерно поглощение в интервалах 2800-2960 см-1 (валентные колебания связей С-Н) и 1360-1480 см-1 (деформационные колебания связей групп СН2 и СН3). Химические свойства алканов Высокая прочность С-С и С-Н связей в алканах обусловливают их высокую инертность, а низкая полярность и поляризуемость – склонность к гомолитическим реакциям. По этим причинам большинство реакций алканов имеет радикальный характер, а для успеха их протекания требуется внешнее энергетическое воздействие (например, температура, фотолиз, катализ). Свободнорадикальные реакции алканов 1. Галогенирование. Алканы очень активно взаимодействуют с фтором, реакция с хлором происходит при освещении и нагревании, взаимодействие с бромом осуществляют при совместном освещении и нагревании. Такие различия обусловлены различной энергетикой реакций галогенирования. Механизм развития цепей этих реакций можно представить двумя последовательными стадиями. X + RH HX + R (1) R + X2 RX + X (2) При достаточно длинных кинетических цепях стехиометрия брутто-реакции определяется реакциями (1) и (2). Суммирование левых и правых частей уравнений этих реакций приводит к стехиометрическому уравнению. RH + X2 RX + HX Фторирование алканов В случае фторирования реакция протекает самопроизвольно со взрывом из-за высокого теплового эффекта каждой стадии и быстроты их протекания. Инициирование осуществляется посредством бимолекулярной реакции F2 + RH RF + F + R эндотермичность которой не превышает 20 кДж/моль. Это обеспечивает умеренное cкорoсти инициирования. При этом высокая экзотермичность реакций (1) и (2) обусловливает близкие к нулю их энергии активации и высокие скорости. В целом это приводит к столь высоким скоростям брутто-реакции и соответственно скоростям тепловыделений, что большая часть выделяющегося тепла идет на разогрев реакционной смеси, приводящего в конечном счете к тепловому взрыву. На практике, чтобы осуществить фторирование с контролируемыми скоростями исходные реагенты разбавляют инертными газами (N2, Ar и др). Хлорирование алканов Расщепление молекулы хлора на хлоррадикалы Cl2 + M M + 2Cl требует подвода энергии, равной энергии связи Cl-Cl 243 кДж/моль. Поэтому в обычных условиях реакция хлорирования не идет. Более эффективное инициирование можно осуществить путем: а) Повышения температуры б) Фотооблучения реакционной смеси УФ-светом. Cl2 + h 2Cl в) Проведения реакции в присутствии иницииаторов – соединений, легко диссоциирующих на свободные радикалы, которые дают начало цепному процессу. In2 2In In + Cl2 InCl + Cl В качестве инициаторов хлорирования и других свободно радикальных реакций можно использовать органические пероксиды и азосоединения, например бензоилпероксид
или азобисизобутиронитрил
Любой из способов инициирования более эффективно генерирует свободные радикалы, благодаря чему их концентрация становится достаточной, чтобы обеспечить высокие скорости брутто-реакций. Реакции развития цепей (1) и (2) в случае хлорирования метана имеют тепловые эффекты соответственно –4, 2 и –109 кДж/моль. Экзотермичность обоих стадий обусловливает низкие энергетические барьеры реакции (1) и (2), что при эффективном инициировании обеспечивает достаточно высокие скорости реакций хлорирования. Бромирование алканов Стадия развития цепей (1) для реакции бромирования метана умеренно эндотермична (+67 кДж/моль). Это обусловливает ее высокий энергетический барьер и обратимость. Как следствие длины кинетических цепей низки (от нескольких единиц до 2-3 десятков). Поэтому бромирование метана и других мало реакционноспособных субстратов протекает трудно, более эффективно реакция протекает с реакционноспособными субстратами. Иодирование алканов Реакция иодирования алканов оказывается невозможной, так как стадия (1) высоко эндотермична (+138 кДж/моль). Это обусловливает смещение равновесия этой реакции нацело в левую сторону. Наоборот, известно, что йодалканы под действием НJ легко превращаются в алканы и йод. Наибольший практический интерес имеют реакция хлорирования алканов и их производных. На самом деле реакция нитрования осложняется деструкцией углеродной цепи с образованием побочных низкомолекулярных нитроалканов и продуктов окисления. 4. Реакции окисления. Реакции окисления алканов осуществляет в газовой и жидкой фазах. Из газофазных реакций практическое значение имеет окисление метана. Окисление метана имеет последовательно–параллельный характер в соответствии со схемой:
Процесс осуществляют с целью промышленного синтеза метанола и формальдегида. Различают термическое и каталитическое окисление. Первое осуществляют при температурах 500-600о, второе – при 350-450о и катализе AlPO4. Жидкофазное окисление имеет практическое значение для синтеза высших карбоновых кислот, являющихся сырьем для получения ПАВ:
Эти процессы проводят при 120-150оС при инициировании солями Со или Mn, например Co+3 + RH Co+2 + R + H+ Образующиеся радикалы R дают начало цепному процессу, который представляет собой совокупность цепей свободнорадикальных реакций с вырожденным разветвлением цепей. Ключевую роль в этом процессе играют образование -кетогидропероксида, который вовлекается в реакции деструктивного окисления с образованием карбоновых кислот.
5. Реакции крекинга. При нагревании выше 500оС алканы подвергаются пиролитическому разложению с образованием сложных смеси продуктов, соотношение которых зависит от температуры и времени реакции. Вся совокупность протекающих при этом реакций называется крекингом. Это типичный свободнорадикальный процесс, инициируемый гомолитическим распадом углерод-углеродных связей исходных алканов. Механизм этого процесса может быть представлен на примере крекинга н-бутана: Инициирование
Развитие цепей
В соответствии с приведенным механизмом основными продуктами крекинга являются алканы и алкены, содержащие меньшее число углеродных атомов, чем исходный алкан. Если крекинг проводить более глубоко и в жестких условиях, то такой исчерпывающий процесс приводит к преимущественному образованию этилена. Этот процесс называется пиролизом. Он реализуется в широких масштабах для производства этилена. Другим важным процессом превращения алканов является каталитический крекинг, осуществляемый на алюмосиликатных катализаторах при 400-450оС. Изомеризующее и ароматизирующее действие катализаторов в этом процессе обусловливает образование в значительных количествах алканов с разветвленной структурой и арены. Благодаря этому образующиеся углеводородные фракции имеют высокое октановое число. Поэтому каталитическийкрекинг является основным процессом для промышленного производства высокооктановых бензинов. Особое место среди процессов крекинга занимает термический крекинг метана, осуществляемый для промышленного синтеза ацетилена. CH4 С целью обеспечения необходимой температуры процесса его совмещают с процессом сжигания метана в едином реакционном объеме.
Реакция гидрирования Реакции гидрирования алкенов являются иллюстрацией ненасыщенного характера алкенов в силу отмеченной выше слабости -связи. Последняя легко рвется под действием гидрирующего агента и по освобождающимся валентностям присоединяются атомы водорода.
В качестве катализаторов используют Pd, Pt, Ni, нанесенные на пористые носители: -AI2O3, SiO2, алюмосиликаты, активированный уголь. Гидроборирование алкенов. При взаимодействии диборана или замещенных диборанов с алкенами образуются триалкилпроизводные бора 6RCH=CH2 + B2H6 2[RCH2CH2]3B 4RCH=CH2 + B2H4(C2H5)2 2(RCH2CH2)2B(C2H5) Реакция представляет собой электрофильное присоединение BH3 или С2Н5 BH2 к двойной связи, причем электрофильным центром является атом бора со своей незаполненной орбиталью. Полученные борорганические соединения являются важными промежуточными продуктами при синтезе соответствующих соединений. Так, их гидролитическое расщепление в присутствии кислот приводит к образованию соответствующих алканов.
Обработка триалкилборана пероксидлом водорода при катализе основаниями приводит к образованию спиртов, причем конечным результатом является присоединение воды к алкену против правила Мaрковникова:
Реакции циклоприсоединения. Алкены способны вступать в реакции, в которых образуется цикл. Такие реакции называются циклоприсоединением. Эти реакции подразделяются в зависимости от числа атомов, участвующих в образовании цикла (цифры в квадратных скобках) a) Циклоприсоединение [2+1]. К этому типу относятся реакции алкенов с карбенами.
б) Циклоприсоединение [2+2].
в) Циклоприсоединение [2+4].
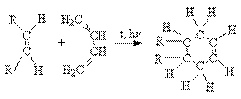
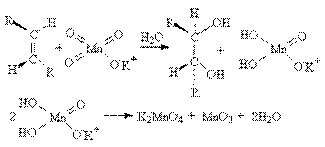
Реакции окисления алкенов. а) Окисление разбавленными растворами KMnO4 в слабощелочной среде (окисление по Вагнеру).
Эта реакция протекает стереоселективно как цис-присоединение двух гидроксильных групп:
б) Окисление концентрированными растворами окислителей. (KMnO4, CrO3, HNO3) При действии этих окислителей происходит разрыв молекулы алкена по месту двойной связи с образованием кетонов и кислот.
Эта реакция широко используется для определения строения алкенов. Реакции полимеризации. а) Катионная полимеризация. Выше отмечалось, что если в качестве нуклеофильного реагента по отношению к двойной связи выступает карбкатион, то это открывает возможность роста его цепи за счет многократно повторяющихся актов присоединения растущего карбкатиона к двойной связи. такой процесс называется катионной полимеризацией. Катионная полимеризация катализируется протонными кислотами, например, серной или органическими сульфокислотами. Механизм этой реакции может быть представлен на примере полимеризации изобутилена: Инициирование:
Рост цепи:
Можно видеть, что роль кислоты - катализатора заключается в генерировании исходных карбкатионов, выступающих в роли электрофильных агентов по отношению к двойной связи. Предпосылкой катионной полимеризации является разветвленная структура алкена, обусловливающая образование стабильных третичных карбкатионов. б) радикальная полимеризация В присутствии генераторов активных свободных радикалов алкены полимеризуются по свободнорадикальному механизму: Инициирование:
Развитие цепей:
Обрыв цепей: где в качестве инициатора выступают органические пероксиды, гидропероксиды, азобисизобутиронитрил. Поэтому полимеризация наиболее инертного этилена осуществляется в наиболее жестких условиях (200оС, давления до 150 мПа). В этих условиях инициирование осуществляется кислородом, который являясь бирадикалом обладает достаточной активностью при высоких температурах для генерирования радикальных активных центров, например:
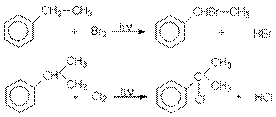
Особенности этих реакций - регулярное строение полимеров, обусловливающее их Методы получения 1. Заместительное галогенирование. Объектом заместительного галогенирования могут быть алканы. В общем виде реакции заместительного галогенирования алканов выражаются уравнением: RH + Hal2 RHal + HНal Таким способом можно получить фтор-, бром- и хлоралканы. Подробно об особенностях этих реакций – см. Химические свойства алканов. Аллильное хлорирование алканов можно осуществить при высоких температурах (400-500оС) в паровой фазе
Для аллильного бромирования алканов в качестве реагента используют бромсукцинимид
Хлорирование и бромирование боковых цепей алкиларенов можно осуществлять в условиях инициирования свободнорадикальных реакций (химические инициаторы, фотоинициирование), причем наиболее реакционноспособным оказывается -положения боковой цепи:
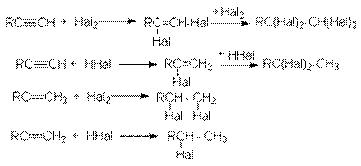
2. Присоединительное галогенирование алкенов и алкинов. Возможности этого метода можно проиллюстрировать следующими реакциями:
Особенности приведенных реакций подробно обсуждались в разделах, посвященных химическим свойствам алкенов и алкинов. Здесь лишь отметим, что при реализации этих реакций в промышленности с целью форсирования процессов используют катализаторы типа кислот Льюиса (FeCl3, SnCl4, SbCl3 и др.) 3. Реакции замещения гидроксильных групп на галоген в спиртах. К таким реакциям относятся: а) взаимодействие галогеноводородов со спиртами
в) Взаимодействие галогенангидридов кислот (PСl5, PBr5, SF4) c альдегидами и кетонами
где R’=H, R, Ar. 4. Замещение галогена на галоген. Замещение более легкого галогена в галогенуглеводороде более тяжелым RCl + KBr RВr + KCl обеспечивается проведением реакции при умеренных температурах и избытке галогенирующего реагента в неполярных апротонных растворителях. Замещение более тяжелого галогена более легким RJ + KCl RCl + KJ обеспечивается проведением реакции при повышенных температурах и избытке галогенирующего реагента в высокополярной среде в присутствии катализатров - кислот Люиса, например, Сu+, Ag+ и пр. Химические свойства Химическое поведение галогенуглеводородов определяется такими факторами как энергия связи С–Hal, полярность этой связи и ее поляризуемость. Так, относительная слабость связей С–Cl, C–Br и СJ обусловливает их предпочтительное гомолитическое расщепление по сравнению со связями С–С и С–Н. В то же время полярность связей С–Hal и их более высокая поляризуемость по сравнению со связями С–С и С–Н является предпосылкой для их гетеролитического расщепления. Приводимые ниже реакции являются иллюстрацией этих положений. 1. Замещение галогена на водород. Восстановление галогенпроизводных до углеводородов осуществляется водородом в присутствии обычных катализаторов гидрирования (Ni, Pt, Pd):
Реакция имеет гомолитический характер, связанный с предварительной сорбцией реагентов на поверхности катализатора, причем водород претерпевает диссоциативную адсорбцию с вовлечением адсорбированных атомов водорода в реакцию восстановления Реакция (1) имеет важное практическое значение как метод переработки галогенорганических отходов в промышленном органическом синтезе. В отличие от другого метода обезвреживания галогенорганических отходов, сжигания, этот метод является ресурсосберегающим и экологически безопасным, так как при его реализации регенерируется углеводородная составляющая исходного сырья и исключается образование высокотоксичных полихлордибензодиоксинов и полихлордибензофуранов. Другой метод замещения галогена на водород – взаимодействие галогенпроизводных углеводородов с иодоводородной кислотой при нагревании
Метод имеет препаративное значение. 2. Взаимодействие с металлами. а) реакция димеризации (синтез Вюрца) 2RHal + Na R–R + 2NaHal Такой характер взаимодействия является выражением природы реагентов. Натрий как очень активный металл легко отдает свой электрон молекуле галогенуглеводорода, которая выступает в роли слабого электрофилаРеакция имеет препаративное значение. б) Взаимодействие с магнием (реакция Гриньяра).
Механизм этой реакции в части ее инициирования подобен механизму реакции Вюрца
3. Реакции нуклеофильного замещения Реакции нуклеофильного замещения – наиболее типичный круг реакций, в которых галогенуглеводороды выступают в качестве субстратов. Результатом этих реакций является замещение галогена на другой атом или группу, которые либо непосредственно выступают в роли нуклеофильного реагента, либо входят в его состав в качестве фрагмента. Наиболее типичными реакциями нуклеофильного замещения галогеналканов и других галогенпроизводных являются: а) реакции гидролиза RHal + H2O ROH + HHal RHal + NaOH ROH + NaHal б) реакции образования простых эфиров (реакция Вильямсона) RHal + R`ONa ROR` + NaHal в) синтез сложных эфиров R1Hal + RCOONa RCOOR1 + NaHal г) аммонолиз RHal + 2NH3 RNH2 + NH4Hal д) синтез нитросоединений и нитритов е) синтез нитрилов и изонитрилов ж) синтез тиолов и сульфидов R–Hal + NaSH RSH + NaHal R–Hal + R1SNa RSR1 + NaHal 2R–Hal + Na2S RSR + 2NaHal з) синтез фосфорорганических соединений (CH3)2PH + CH3CH2Br + NaOH (CH3)2PCH2CH3 + NaBr + H2O диметилэтилфосфин и) синтез углеводородов на основе Mg-органических соединений R–MgHal + R’Hal R–R’ + MgHal2 к) замещение галогена на галоген RX + NaY RY + NaY (см. подробно методы получения галогеналканов) Из приведенных реакций можно видеть, что реакции нуклеофильного замещения могут служить методом образования связей С–С, С–О, С–S, C–N, C–P и C–Hal. Алканолы (спирты) Алканолы - результат замещения атомов водорода в алканах на ОН - группы. Различают одноатомные и многоатомные алканолы. Последние представляют собой результат замещения атомов водорода в алканах при различных углеродных атомах, например:
Эмпирическая формула одноатомных спиртов СnH2n+2O. Методы получения 1. Гидратация алкенов
2. Гидроборирование алкенов
3. Гидролиз галогеналканов 2RCl + Na2CO3 +H2O 2ROH + 2CO2 + 2NaCl 4. Гидрирование карбонильных соединений. |
Последнее изменение этой страницы: 2017-04-12; Просмотров: 1022; Нарушение авторского права страницы

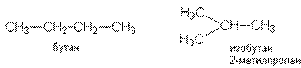

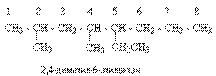


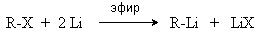



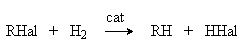








 CH2=CH2 + CH CH + H2
CH2=CH2 + CH CH + H2