
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Коллоидные системы и предмет коллоидной химииСтр 1 из 29Следующая ⇒
Коллоидная химия
Коллоидные системы и предмет коллоидной химии Коллоидные системы
Историческая справка Первоначально коллоидная химия была лишь главой физической химии. Теперь это самостоятельная дисциплина со своим кругом идей. Были разработаны специальные специфические коллоидно-химические методы исследования: ультрамикроскопия, электронная микроскопия, ультрацентрифугирование, электрофорез и т.д. Практика показала огромное значение коллоидной химии для современной техники. Невозможно указать отрасль народного хозяйства, в которой не использовались бы коллоидные системы и коллоидные процессы. С коллоидными системами человек имел дело с незапамятных времен. Однако изучение их началось сравнительно недавно. Обычно считают, что основателем коллоидной химии является английский ученый Томас Грэм (*)(1805-1869), который в 50-60-е годы позапрошлого столетия ввел в обращение основные коллоидно-химические понятия. Однако не следует забывать, что у него имелись предшественники, и прежде всего – Яков Берцелиус(*) и итальянский химик Франческо Сельми(*). В 30-е годы XIX века Берцелиус описал ряд осадков, проходящих при промывании через фильтр (кремниевая и ванадиевая кислоты, хлористое серебро, берлинская лазурь и др.). Эти проходящие через фильтр осадки Берцелиус назвал «растворами», но в то же время он указал на их близкое сродство с эмульсиями и суспензиями, со свойствами которых он был хорошо знаком. Франческо Сельми в 50-е годы XIX века продолжил работы в этом направлении, ища физико-химические различия между системами, образованными осадками, проходящими через фильтр (он назвал их «псевдорастворами») и обычными истинными растворами. Английский ученый Майкл Фарадей(*) в 1857 г. синтезировал коллоидные растворы золота – взвесь Au в воде размерами частиц от 1 до 10 нм. и разработал методы их стабилизации. Эти «псевдорастворы» рассеивают свет, растворенные в них вещества выпадают в осадок при добавлении небольших количеств солей, переход вещества в раствор и осаждение из него не сопровождаются изменением температуры и объема системы, что обычно наблюдается при растворении кристаллических веществ. Томас Грэм развил эти представления о различии между «псевдорастворами» и истинными растворами и ввел понятие «коллоид». Грэм обнаружил, что вещества, способные к образованию студнеобразных аморфных осадков, такие как гидроокись алюминия, альбумин, желатина, диффундируют в воде с малой скоростью по сравнению с кристаллическими веществами (NaCl, сахароза). В то же время кристаллические вещества легко проходят в растворе через пергаментные оболочки («диализируют»), а студнеобразные вещества не проходят через эти оболочки. Принимая клей за типичный представитель студнеобразных не диффундирующих и не диализирующих веществ, Грэм дал им общее название «коллоид», т.е. клееобразный (от греческого слова колла – клей). Кристаллические вещества и вещества, хороши диффундирующие и диализирующие он назвал «кристаллоидами».
Перечислим аномальные свойства некоторых растворов, которые мы называем теперь коллоидными системами. Свойства коллоидных систем: 1. рассеивание света (опалесценция) (указывает на неоднородность, многофазность системы). Опалесценция становится особенно заметной, если, как это делал Тиндаль (*) через коллоидный раствор пропускать пучок сходящихся лучей, поставив между источником света и кюветой с раствором линзу. При этом растворы, прозрачные в проходящем свете, в боковом освещении проявляют все свойства мутных сред. В коллоидной жидкости, наблюдаемой сбоку, образуется яркий светящийся конус (конус Тиндаля).
2. медленная диффузия 3. малое осмотическое давление (пп. 2 и 3 говорят о наличии в системе крупных частиц) 4. коллоидные растворы способны к диализу, т.е. с помощью мембраны могут быть отделены от примесей 5. способны к коагуляции (разрушению) системы при: добавлении примесей, изменении Т, перемешивании и т.д. 6. иногда обнаруживают явление электрофореза, открытое Рейссом(6) в России в 1808 г., т.е. частицы в системе могут обладать зарядом.
Чтобы представить, чем занимается наука «Коллоидная химия», надо ответить на вопрос, что такое коллоиды или коллоидные системы? Предмет коллоидной химии Коллоидная химия – наука о поверхностных явлениях и дисперсных системах.
К поверхностным явлениям относятся процессы, идущие на границе раздела фаз, в межфазном поверхностном слое и возникающие в результате взаимодействия сопряженных фаз. Напомним, что фазой называется часть термодинамической системы, обладающая определенными физическими и химическими свойствами и отделенная от других частей системы поверхностью раздела. В истинных растворах вещество раздроблено до молекулярного состояния и между растворенным веществом и растворителем нет границы раздела. Причиной поверхностных явлений является существование на границе раздела контактирующих фаз ненасыщенного поля межатомных, межмолекулярных сил, которое возникает из-за разного состава и строения соприкасающихся фаз и различия в связях их поверхностных атомов и молекул.
Поверхностные слои жидких и твердых тел, прилегающих к поверхности раздела фаз, резко отличаются по многим физико-химическим показателям от свойств фаз в глубине их объема (уд.энергия, плотность, вязкость, уд. электропроводность и др.). Отличия связаны и c определенной ориентацией молекул в поверхностных слоях и иным энергетическим состоянием их в сравнении с молекулами в объеме. Кроме того, в многокомпонентных системах (растворах) состав поверхностного слоя не совпадает с составом объемных фаз. Особенности поверхностных слоев обусловлены наличием избытка поверхностной энергии. Свойства поверхности раздела тем сильнее влияют на поведение системы в целом, чем больше площадь поверхности (Sуд). Этим объясняется доминирующая роль поверхностных явлений в свойствах высокодисперсных систем, Sуд которых достигает огромных величин. Наличие избыточной энергии в поверхностном слое молекул обусловлено неполной скомпенсированностью межмолекулярных сил притяжения у молекул поверхностного слоя вследствие их слабого взаимодействия с граничащей фазой.
Коллоидная химия изучает дисперсные системы – гетерогенные системы, состоящие из двух и более фаз, одна из которых дисперсная фаза – раздроблена (прерывна), а другая - дисперсионная среда - является непрерывной частью системы.
Положение о микрогетерогенной природе коллоидных растворов и других дисперсных систем имеет фундаментальное значение. За его открытие австрийский ученый Зигмонди(*) стал лауреатом Нобелевской премии по химии в 1925 г. Выделение в особую группу дисперсных частиц вызвано отличием их по физическим и химическим свойствам от аналогичных свойств крупных объектов одного и того же вещества. К числу таких свойств относятся прочность, теплоемкость, Тпл, магнитные и электрические характеристики, реакционная способность. Эти различия вызваны размерными эффектами. Особые свойства выражены тем сильнее, чем меньше размер частиц, особенно это проявляется у наночастиц. Эти свойства открывают принципиально новые практические приложения химии, физики, биологии. Изучение свойств дисперсных частиц (методов получения, структуры, физики и химии) относятся к наиболее актуальным и перспективным задачам ряда дисциплин.
Дисперсные частицы могут иметь самую различную форму: цилиндрическую, сферическую, прямоугольную, неправильную. Например, к дисперсным частицам относятся: системы с частицами кубической, шарообразной формой – золи, эмульсии, взвеси, пасты; нитевидные – волокна нервных клеток, 2-х - мерные мышечные волокна, капилляры, поры (древесина, ткани, волосы, кожа), плёнки – поверхностные слои на границах раздела в эмульсиях, пенах, в порах катализаторов и адсорбентов, мембран.
Таким образом, 1 м3 исходного вещества можно раздробить на кубики с длиной ребра а, вытянуть в нить с сечением а или расплющить в пленку толщиной а. Если частицы имеют неправильную форму, то для использования понятия «поперечный размер», их форму приравнивают к сферической с эквивалентным диаметром.
Количественные характеристики дисперсной системы: 1. Размер частиц dср, dmin, dmax 2. Концентрация частиц ν = nd/V, где nd – число частиц дисперсной фазы в единице объема дисперсионной среды V 3. Раздробленность системы характеризуется дисперсностью D и величиной удельной поверхности дисперсной фазы Sуд: Первый вариант количественной оценки - основной D= 1/d и Sуд = S /V, (1.1) где d – минимальный размер частицы, S – суммарная площадь межфазной поверхности, V- объем тела. Например, у частицы кубической формы с размером ребра d = 10-8м Sуд = 6 d2/ d3= 6/ d = 6 *108м-1 Для нити сечением d2= 10-8 * 10-8 Sуд = 4* 108м-1 Для пластины толщиной d= 10-8 м Sуд = 2 *108м-1
Для систем, содержащих частицы сферической формы с радиусом r Sуд = 4 π r2 / 4/3 π r3 = 3/ r Второй вариант ( в учебнике МГУ — Щукина ): D= S /V, (1.2) где S – суммарная площадь межфазной поверхности, V - объем тела, Sуд = S /∑ m = D / ρ, где ρ = плотность данного вещества.
Итак, коллоидные системы имеют два характерных признака: 1. гетерогенность 2 дисперсность.
Безусловно, первый из них имеет превалирующее значение для коллоидных систем, поскольку в отсутствие границы раздела фаз поверхностные явления не возникают.
Межмолекулярные связи
Межмолекулярное взаимодействие может иметь различный характер: 1. химические связи – образуются путем перекрывания электронных орбиталей и потому сугубо специфичны 2. водородные связи возникают между молекулами, содержащими функциональную группу – ОН: кислотами, щелочами, водой и др. веществ -
силикагель - Si – OH H O O - Si – OH H
3. Силы Ван-дер-Ваальса(*) (молекулярные связи), действующие между любыми молекулами.
Отличия молекулярных сил притяжения от химических: а) по величине энергии: Емол= 5 - 50 кДж/моль Е хим = 80 – 800 кДж/моль б) молекулярные силы неспецифичны в) различие по радиусу действия r хим~ 10-8 r мол > 10-7 см г) молекулярные силы аддитивны, а химические связи насыщаемы
Молекулярные силы взаимодействия включают в себя: а) ориентационные силы (Кеезома) (*) Возникают между полярными молекулами в результате взаимодействия дипольных моментов. Из-за диполь-дипольного взаимодействия молекулы приобретают определенную ориентацию относительно друг друга
Энергия ориентационного взаимодействия сильно зависит от расстояния между молекулами: Е μ ˉ 1/ μ ˉ 2 = - А1/r6 (2.1)
У полярной молекулы – центр тяжести «+» и «-» зарядов не совпадает (дипольный момент μ ˉ i ≠ 0). Степень полярности зависит от дипольного момента μ ˉ i. Полярность многоатомных молекул определяется полярностями отдельных связей и их расположением относительно друг друга. К неполярным неорганическим веществам относятся: элементы, симметрические молекулы газов, некоторые соли (сульфиды металлов). При рассмотрении полярности или неполярности молекул органических следует обращать внимание не только на наличие полярной группы в молекуле, но и на расположение их в структурной формуле молекулы. Например:
б) индукционные силы (силы Дебая(*)). Диполь у неполярной молекулы возникает в электрическом поле или под действием поля полярной молекулы. Энергия связи зависит от поляризуемости молекул и также сильно уменьшается с увеличением расстояния: Е инд = - А2/r6 (2.2)
в) дисперсионные силы (силы Лондона(*)) действуют между всеми молекулами. Возникают из-за непрерывного движения электронов в атомах, приводящего к образованию мгновенных диполей. В свою очередь электрическое поле мгновенного диполя одного атома индуцирует дипольный момент соседнего, что приводит к возникновению сил притяжения. Е дисп = - А3/r6 (2.3)
где α – поляризуемость молекулы. Дисперсионные силы притяжения не зависят от температуры. Молекулярные силы быстро убывают с увеличением расстояния между молекулами. Рассмотрим соотношения между отдельными силами притяжения молекул (табл. 2.1): Таблица 2.1 Определения поверхностного натяжения Возьмем объект такой конфигурации, чтобы при его разрыве плоскостью скольжения образовались две составляющих части с площадью поверхности S. При разрыве тела тратится определенная работа, идущая на разрыв межмолекулярных сил. Естественно, что эта работа пропорциональна площади межфазной поверхности: W=σ S (2.7)
+
Рис.2.2. К определению поверхностного натяжения как работы образования единицы поверхности
На новой поверхности образуется слой молекул, обладающих большей энергией, чем молекулы внутри фазы. Коэффициент пропорциональности между работой и площадью межфазной поверхности называется коэффициентом поверхностного натяжения или просто поверхностным натяжением .
Исходя из приведенного уравнения, виден физический смысл поверхностного натяжения как работы: 1. Поверхностное натяжение численно равно работе обратимого изотермического образования единицы поверхности Понятие обратимого процесса накладывает определенное ограничение на использование этого определения, так как не всякую границу раздела фаз можно получить приведёнными рассуждениями обратимо. Например, получение новой площади границы раздела т/г невозможно получить обратимо, т.к. реально нужно учитывать необратимую деформацию молекул. Поэтому часто используют определение поверхностного натяжения как удельной поверхностной энергии.
2. Поверхность раздела фаз обладает избытком нескомпенсированной энергии. Этот избыток в расчете на единицу поверхности составляет удельную свободную поверхностную энергию . Для увеличения площади жидкой фазы нужно преодолеть внутреннее давление и совершить определенную механическую работу. Если увеличение площади производится при Р, Т = cоnst или V, T = cоnst, то оно сопровождается увеличением поверхностной энергии системы.
Термодинамическое определение поверхностного натяжения вытекает из объединенного уравнения I и II начал термодинамики. Запишем его для гетерогенной системы относительно внутренней энергии U: dU = TdS – PdV +σ dS +∑ μ idni +φ dq (2.8) при S, V, ni, и q = cоnst dU = σ dS (2.9)
Отсюда получаем, т.е. поверхностное натяжение – частная производная от внутренней энергии по площади поверхности раздела фаз при постоянных энтропии, объеме, числе моль вещества и заряде поверхности. Так как объединенное уравнение может быть записано относительно и других термодинамических потенциалов, то при соответствующих постоянных параметрах получаем:
Поскольку чаще всего мы имеем дело с процессами, происходящими в изобарно-изотермических условиях, то можно встретить такое определение: Поверхностное натяжение σ – это избыточная удельная поверхностная энергия Гиббса (*). Для индивидуальных веществ это определение достаточно строгое. Для единицы поверхности можно записать: σ = Gs (2.12) «Избыточность» означает, что энергия поверхностных молекул жидкости больше энергии молекул в ее внутреннем объеме. 3) Поверхностное натяжение помимо энергетического (термодинамического) физического смысла имеет и силовой (механический). Это может прояснить простой опыт: В l С
А D dx A׳ D ׳ G
Fֿ Рис.2.3. Рамка Дюпре(*)
На проволочной рамке помещается подвижная перекладина АD длиной l, легко скользящая по рамке. Опускаем рамку в водный раствор мыла. На рамке образуется двухсторонняя мыльная пленка, стягивающая часть рамки длиной l. Приложим к подвижной перекладине АD направленную вниз силу F (груз G). Под действием силы F перекладина АD переместится на бесконечно малое расстояние dx и займет положение А׳ D׳ . Сила F произведет при этом работу dW=Fdx. (2.13) Если T=const, то эта работа затрачивается только на увеличение площади пленки: dS = 2l dx (2.14) dW = σ dS. (2.15) Определим условие силового механического равновесия перекладины АD при приложении силы F: dW = F dx = σ dS = σ 2l dx. (2.16) Такое равновесие обеспечивает сила, направленная в противоположную сторону и равная: σ = F/2l. (2.17) Температура Т Влияние добавок
Поверхностное натяжение растворов отличается от поверхностного натяжения растворителя. Зависимость σ ж/г = f(С) при Т=const называется изотермой поверхностного натяжения. Знак dσ /dс указывает на характер зависимости σ от концентрации С. Условимся рассматривать изотерму поверхностного натяжения только для водных растворов, поэтому при С=0 поверхностное натяжение σ о равно σ н2о при данной температуре.
σ 1
с
Рис.2.6. Изотермы поверхностного натяжения на границе ж/г в зависимости от концентрации растворенного вещества
Для водных растворов различают 3 основных типов изотерм: 1. поверхностно-неактивные вещества, не изменяющие поверхностное натяжение (кривая 1). 2. поверхностно- инактивные вещества (электролиты), которые в воде диссоциируют с образованием ионов, которые хорошо гидратируются, т.к. Еион/н2о> Е н2о/н2о, поэтому ионы интенсивно втягиваются вглубь раствора, dσ /dс > 0 (кривая 2).
Для границы раздела фаз вода - воздух это соли, щелочи, минеральные кислоты, т.е. любые соединения, образующие в растворе только неорганические ионы. Их действие объясняется следующим образом: силы притяжения ионов и диполей воды сильнее, чем диполей друг к другу, поэтому при растворении ПИВ в воде увеличиваются межмолекулярные взаимодействия в поверхностном слое, а, следовательно, увеличивается и σ. Эффект увеличения σ от добавки ПИВ в воду обычно незначителен. Это видно из рис.2.5. Так, поверхностное натяжение чистой воды при 20°С равно 72, 8 мДж/м2, для 1 % раствора NaOH оно составляет 73, 0 мДж/м2 и лишь у 10% раствора NaOH ст достигает 77, 5 мДж/м2.
3. поверхностно-активные вещества, которые уменьшают поверхностное натяжение на границе раздела фаз (кривая 3).
Способность уменьшать поверхностное натяжение называется поверхностной активностью К ПАВ относятся органические молекулы с несимметричным строением молекул, состоящие из полярных и неполярных групп – с дифильным строением ( рис.2.7) :
полярная группа: -СООН; - -NO2; -СHO; - ОН; -NH2; SO2OH неполярный углеводород- ный радикал
Рис. 2.7. Условное изображение молекулы ПАВ Полярные группы в воде гидратируются, неполярная часть молекул ПАВ представляют собой гидрофобную углеводородную цепь или радикал.
Молекула ПАВ из-за своего дифильного строения по-разному взаимодействует с молекулами воды в растворе: полярная часть легко гидратируется (благодаря этому идет растворение молекул ПАВ – этот процесс энергетически очень выгоден), неполярный углеводородный радикал, слабо взаимодействуя с водой, препятствует межмолекулярному взаимодействию диполей воды друг с другом. Ен2о/н2о > Ен2о/ПАВ (напоминаем, что взаимодействие молекул воды друг с другом достаточно сильное – ориентационное, индукционное, дисперсионное, плюс водородные связи), поэтому энергетически выгоднее удалить неполярные длинные углеводородные радикалы из объема.
В результате на поверхности образуется определённым образом ориентированный адсорбционный слой, в котором полярная часть обращена в воду, а неполярный радикал - в контактирующую фазу (например, в воздух). При этом уменьшается избыточная поверхностная энергия, а, следовательно, и поверхностное натяжение.
Кривая 3 на рис. 2.6. характеризует зависимость σ =f(С) для водных растворов полярных органических веществ с не очень длинными цепями и недиссоциирующими или слабодиссоциирующими группами алифатических спиртов, аминов, жирных кислот. Для них поверхностное натяжение падает сначала линейно, затем по логарифмическому закону. Этот тип зависимости σ =f(С) хорошо описывается эмпирическим уравнением Шишковского: σ = σ о – В ln(1+A С). (2.21) Физический смысл коэффициентов А и В мы обсудим несколько позже. (Значение константы А возрастает в 3-3, 5 раза при переходе к гомологу, а В = RTГ∞, где Г∞ — предельная адсорбция)
Обычно не даю, чтобы не путать: Существует большая группа ПАВ с большим гидрофобным радикалом и сильно гидратирующейся полярной группой. В растворах таких соединений с увеличением концентрации до некоторой критической величины – ККМ (критической концентрации мицеллообразования) образуются мицеллы – агрегаты из ориентированных молекул ПАВ. Поверхностное натяжение таких растворов определяется индивидуальными молекулами ПАВ, т.к. мицеллы почти не снижают поверхностное натяжение раствора – кривая 4.
2.2.4. Экспериментальные методы определения поверхностного натяжения
Основная характеристика свойств поверхности раздела фаз – удельная свободная поверхностная энергия и численно равная ей величина поверхностного натяжения могут быть сравнительно легко и с большой точностью определены для легкоподвижных границ раздела – ж/г и ж1/ж2. Существует большое число методов определения поверхностного натяжения. Остановимся на общих принципах основных методов измерения σ на границе ж/г. Для измерения поверхностного натяжения индивидуальных жидкостей пригодны любые методы. У растворов же результаты измерений поверхностного натяжения разными методами могут сильно отличаться из-за медленного установления равновесного распределения растворённых веществ между только что образованной поверхностью и объёмом раствора. Для правильного выбора метода исследования необходимо учитывать кинетику установления равновесных значений поверхностного натяжения. Например, диффузия молекул ПАВ к поверхности раздела фаз происходит достаточно медленно, за короткое время замера не успевает установиться их равновесная поверхностная концентрация, при этом молекулы не успевают должным образом ориентироваться в поверхностном слое. Поэтому для измерений σ в этом случае следует использовать статические или полустатическиме методы, но не динамические. Рассмотрим некоторые наиболее распространенные статические и полустатические методы определения поверхностного натяжения.
1. Статические – основаны на изучении устойчивого равновесного состояния, к которому самопроизвольно приходит система. К ним относятся методы: уравновешивания пластинки, капиллярного поднятия, лежачей или висячей капли.
Метод Вильгельми (*) (метод уравновешивания пластинки). Тонкую пластинку толщиной d, закрепленную на коромысле весов, погружают в исследуемую жидкость, которая хорошо смачивает ее поверхность. На поверхности пластинки образуются мениски. Форма их поверхности и максимальная высота поднятия жидкости определяется уравнением Лапласа.
Вес пластинки определяется статически и при отрыве от поверхности. Суммарный вес жидкости (а, следовательно, сила F, которую надо приложить для уравновешивания пластинки), приходящийся на единицу параметра пластинки, не зависит от формы мениска и при θ =0 равен поверхностному натяжению:
Метод капиллярного поднятия основан на использовании расчетов поверхностного натяжения по формуле Жюрена(*):
где Н – высота поднятия жидкости в капилляре, ρ и ρ о – плотности жидкости и ее насыщенного пара, θ – краевой угол смачивания, g – ускорение силы тяжести.
Разность давлений, возникающая по обе стороны от поверхности жидкости при её искривлении, называется капиллярным давлением. Если капилляр опустить в жидкость, то за счёт смачивания или несмачивания стенок капилляра образуется мениск, т.е. искривление поверхности жидкости и возникает капиллярное давление. Под его влиянием граница жидкости перемещается до тех пор, пока не установится равновесие между гидростатическим давлением и капиллярным. При этом смачивающая жидкость поднимается, а несмачивающая опускается. Измерения σ производят в приборе, схематически изображённом на рис.2.8. Исследуемую жидкость заливают в широкую трубку (см. схему прибора), далее с помощью катетометра измеряют высоту капиллярного поднятия h. Она зависит от поверхностного натяжения жидкости:
Рис. 2.8. Схема прибора для измерения поверхностного натяжения жидкостей методом капиллярного поднятия: 1 - капилляр, 2 -широкая трубка.
Жидкость в капилляре поднимается вверх, пока гидростатическое давление Р не уравновесится с капиллярным Рσ (Рσ =2σ /r), r=ro/сosθ. Применяют тонкие капилляры, что обеспечивает сферичность мениска, хорошо смачиваемые жидкостью, поэтому можно упростить расчет (угол θ ≈ 0о).
Группа методов (лежачей или висячей капли) основаны на изучении формы капель в поле силы тяжести. В этих случаях ведется учет отклонения их формы от сферической. Этот метод ценен для определения поверхностного натяжения при высоких температурах. В этих случаях капли фотографируют длиннофокусной оптикой либо в рентгеновских лучах.
Сопоставляют результаты измерений геометрических параметров, показывающих степень отклонения поверхности от сферической, с табулированными значениями этих параметров (их получают численным интегрированием уравнения Лапласа(*)), находят величину σ.
2) Полустатические методы основаны на изучении условий, при которых система теряет равновесие. Адсорбция 2.4.1. Основные понятия и определения
Адсорбция – процесс самопроизвольного изменения концентрации (перераспределения) компонентов системы между поверхностным слоем и объемной фазой. Более плотная фаза называется адсорбентом (в жидком или твердом агрегатном состоянии). Вещество, которое адсорбируется, называется адсорбат или адсорбтив. Обратный процесс называется десорбцие й.
Для количественного описания адсорбции используются две величины: 1. Абсолютная адсорбция А – число моль или г адсорбата, приходящееся на единицу поверхности или массы адсорбента.
Единицами измерения А являются моль/м2, моль/г или моль/см3. Экспериментально А определяют весовым методом (например, на весах Мак-Бена) при изучении адсорбции из газовой фазы на твёрдом адсорбенте. Увеличение массы (пересчитанное в моль) адсорбента, подвешенного на весах, равно именно А.
2. Избыточная адсорбция (гиббсова) Г – избыток адсорбата в поверхностном слое по сравнению с его количеством в таком же объеме фазы, приходящийся на единицу поверхности или массы адсорбента. Измеряют избыточную адсорбцию также в моль/м2, моль/г или моль/см3. Экспериментально Г определяют по разности концентраций адсорбата в растворе до и после адсорбции (как это делается в лабораторном практикуме).
По своему физическому смыслу А всегда положительна (А > 0). Значение же Г может быть как положительным (вещество концентрируется на поверхности), так и отрицательным (вещество избегает поверхности, как в случае адсорбции ПИВ). По определению А всегда больше Г, но при малых концентрациях адсорбата (можно пренебречь количеством вещества в слое фазы по сравнению с количеством его у поверхности) и сильной его адсорбции А » Г. Обычно это наблюдается в водных растворах ПАВ.
Установлен ряд приближенных критериев, совокупность которых позволяет на основании экспериментальных данных различить физическую и химическую адсорбции. 1. Физическая адсорбция происходит под влиянием сил Ван-дер-Ваальса и по своей природе аналогична процессам конденсации паров адсорбата. Поэтому теплота её близка к теплотам конденсации и составляет –(5 – 40) кДж/моль. Теплота хемосорбции соизмерима с теплотами химических реакций и составляет обычно –(80 – 400) кДж/моль.
Однако хемосорбция из жидких растворов может сопровождаться выделением теплоты, близкой к теплоте физической адсорбции. Таким образом, если наблюдаемые теплоты адсорбции превышают -80 кДж/моль, то можно с достаточной уверенностью утверждать, что исследуемое явление - хемосорбция. Нельзя, однако, делать вывод о физической природе адсорбции в случае малой величины её теплоты.
2. Температурная область протекания физической адсорбции не может значительно превышать температуру кипения адсорбата при давлении опыта. Так, при атмосферном давлении физическая адсорбция воды ограничена Т≈ 1000С. Хемосорбция же может происходить как при низких, так и при гораздо более высоких температурах.
3. Физическая адсорбция на непористых адсорбентах протекает практически мгновенно, и скорость её слабо зависит от температуры. Хемосорбция, как и любая химическая реакция, протекает через образование активированного комплекса с преодолением энергии активации, т.е. является активированной адсорбцией. Скорость такой адсорбции сильно зависит от температуры (эта зависимость передается уравнением Аррениуса(*)).
Однако бывают случаи, например, при хемосорбции кислорода и водорода на поверхности металлов, когда адсорбция протекает очень быстро и практически без зависимости её скорости от температуры.
4. Однозначным критерием установления природы адсорбции является отсутствие значительной температурной зависимости скорости десорбции. Энергия активации десорбции равна сумме энергии активации адсорбции и теплоты адсорбции. Слабая зависимость скорости десорбции от температуры возможна лишь при малых величинах как энергии активации, так и теплоты адсорбции, а это характерно лишь для физической адсорбции.
5. Физическая адсорбция не специфична: она происходит на любых поверхностях (если температура опыта ниже температуры кипения адсорбата). Благодаря этой особенности физическая адсорбция и может быть использована для измерения общей поверхности твердых тел. В противоположность этому хемосорбция происходит только на тех адсорбентах, с поверхностями которых возможно химическое взаимодействие адсорбата (между ними имеется химическое сродство). 6. Физическая адсорбция может приводить к образованию полимолекулярных пленок (полимолекулярной адсорбции), так как силы взаимодействия в последующих слоях практически не отличаются от сил взаимодействия в первом слое. При хемосорбции химическое взаимодействие требует непосредственного контакта адсорбата с поверхностью и возможность полимолекулярной адсорбции исключается.
Однако количество адсорбированного вещества при хемосорбции может в некоторых случаях превышать однослойное покрытие вследствие проникновения адсорбата на некоторую глубину приповерхностного слоя в междоузлия кристаллической решетки адсорбента. При хемосорбции кислорода на серебре или платине адсорбированное количество может более, чем в 3 раза превышать число атомов кислорода, отвечающее монослойному покрытию поверхности. При этом не образуется объемная фаза оксида. 7. Химическая адсорбция локализована, т.е. на каждом центре адсорбции поверхности может адсорбироваться только одна молекула адсорбата (поверхность можно представить шахматной доской, на каждой клетке которой может находиться только одна фигура). Физическая же адсорбция нелокализована, т.е. в этом случае нет жесткой связи молекул адсорбата и центров адсорбции.
Приведенные критерии, рассматриваемые в отдельности, не всегда позволяют однозначно охарактеризовать тип адсорбции, но примененные совместно обычно позволяют надежно отличать физическую адсорбцию от хемосорбции.
Надо, однако, иметь в виду, что при отсутствии резкой границы между явлениями физического и химического взаимодействия возможна адсорбция, характеризующаяся промежуточными свойствами между физической адсорбцией и хемосорбцией. Часто в литературе можно встретить утверждение, что физическая адсорбция – обратимая, а хемосорбция – необратимая. Оно не корректно: хемосорбция, как и любая химическая реакция, идёт до установления равновесия, когда скорость адсорбции равна скорости десорбции. Термин «необратимая адсорбция» следует использовать лишь в тех случаях, когда химическая природа адсорбирующихся и десорбирующихся молекул различна (молекулы распадаются на фрагменты и при десорбции с поверхности выделяются совсем другие частицы). Так, при десорбции хемосорбированного на платине бензола с поверхности удаляется целый набор углеводородов – от метана до циклогексана. .
В общем случае адсорбция является функцией давления Р (для газов) или концентрации С (для жидких растворов) и температуры, т.е. изображается на плоскости в координатах а = f(P, T) или Г = f(C, T).
Обычно один из параметров поддерживают постоянным и адсорбцию графически изображают в виде следующих кривых (рис.2.12):
1. Изотерма - это зависимость адсорбции от давления газа или от концентрации раствора при постоянной температуре. 2. Изобара - это зависимость адсорбции от температуры при постоянном давлении газа (изопикна - при постоянной концентрации). 3. Изостера - зависимость давления (или концентрации) от температуры при постоянной адсорбции. На практике для граф< Популярное:
|
Последнее изменение этой страницы: 2016-03-25; Просмотров: 2648; Нарушение авторского права страницы
 точнее
точнее  , (2.4)
, (2.4)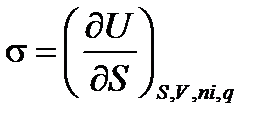 , (2.10)
, (2.10)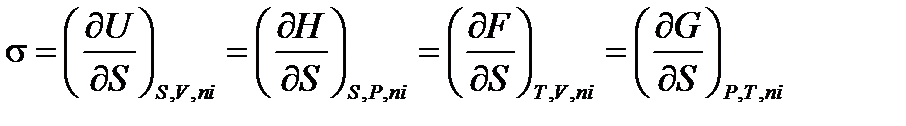 (2.11)
(2.11)
 (2.20)
(2.20) , (2.22)
, (2.22)
 , (2.23)
, (2.23)