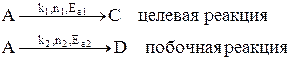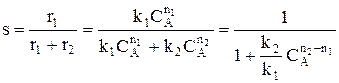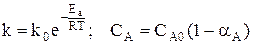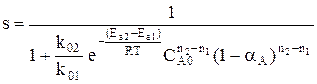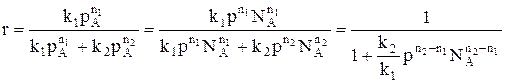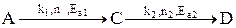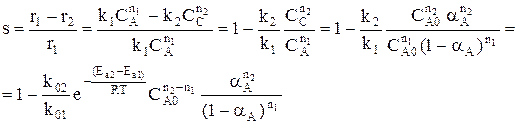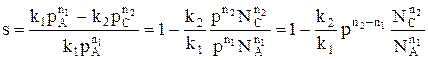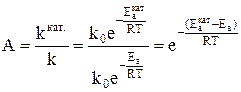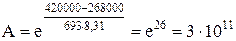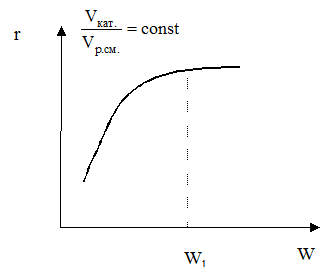|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Закономерности управления простым обратимым гомогенным процессом
Обратимыми называются процессы, в которых одновременно протекают прямая и обратная реакции. Рассмотрим реакцию aA + bB ↔ cC + dD. В начальный момент времени скорость прямой реакции, которая пропорциональна произведению концентраций реагентов А и В
больше скорости обратной реакции, пропорциональной произведению концентраций продуктов С и D
Но с течением времени, по мере увеличения концентрации веществ C и D скорость обратной реакции увеличивается, а скорость прямой реакции уменьшается. В некоторый момент времени они становятся равными
Для газофазных реакций константу равновесия можно выразить через парциальные давления участников реакции, которые пропорциональны их концентрации pi = Ci RT.
Между этими константами существует связь Константу равновесия газофазной реакции можно также выразить через мольные доли компонентов
Парциальное давление каждого компонента пропорционально мольной доле его в смеси pi = p Ni, где р – общее давление системы, Константы Кс и Кр зависят только от температуры, а константа КN еще и от давления. В случае реальных газовых смесей и растворов концентрации и парциальные давления в выражении констант меняют на активность «а» и фугитивность «f». Итак, пределом протекания обратимых процессов является состояние равновесия, при котором скорости прямой и обратной реакции равны, а концентрации и парциальные давления продуктов и реагентов остаются постоянными во времени. При этом достигается некоторая максимальная в этих условиях степень превращения реагентов – равновесная конверсия α * и равновесный выход целевого продукта -β *. Их величина зависит от степени смещения равновесия в сторону образования целевого продукта. Следовательно, при управлении обратимым процессом важно не только обеспечить высокую скорость процесса, но и создать условия, при которых химическое равновесие смещено в сторону образования целевого продукта. Характеристика модели: 1) Процесс простой – можно описать одним стехиометрическим уравнением аА + bВ ↔ cС + dD, где С – целевой продукт; β = α, так как весь реагент превращается только в целевой продукт. 2) Процесс обратимый – α max = α *≠ 100%.

3) Процесс гомогенный – скорость процесса равна скорости химической реакции. Однако следует помнить, что в обратимых процессах скорость равна разности скорости прямой и обратной реакции Объекты управления: скорость химической реакции и положение равновесия. Инструменты управления: кинетические параметры.
Правила смещения равновесия в обратимых процессах сформулированы химиком Ле-Шателье и состоят в следующем: «Если на систему, находящуюся в равновесии, воздействовать извне, изменяя какое-нибудь из условий, определяющих равновесие, то равновесие смещается в том направлении, в котором эффект воздействия уменьшается». Смещение равновесия происходит только в том случае, когда произведенное воздействие неодинаково влияет на скорость прямой и обратной реакции. Например, катализатор всегда одинаково ускоряет как прямую, так и обратную реакцию, и поэтому не оказывает влияния на положение равновесия. Рассмотрим, как влияет на скорость и положение равновесия изменение таких технологических параметров, как температура, давление, концентрация реагентов. 1) Влияние температуры При повышении температуры скорость химической реакции увеличивается за счет увеличения константы скорости, причем, чем больше энергия активации процесса, тем больше влияние температуры на скорость. Энергия активации эндотермического процесса всегда больше энергии активации обратного экзотермического процесса на величину теплового эффекта реакции. Покажем это на энергетической диаграмме. По оси «y» отложим энергию рассматриваемой системы, а по оси «x» - путь или координату реакции. Пусть реакция взаимодействия А+В является экзотермической, ∆ Н < 0. Тогда энергия системы в результате реакции будет понижаться; энергетический уровень продуктов С и D будет меньше энергетического уровня исходных реагентов. Разница между энергиями конечного и начального состояния есть тепловой эффект процесса ∆ Н. Чтобы молекулы веществ А и В прореагировали, они должны преодолеть некоторый энергетический барьер. На это затрачивается энергия, называемая энергией активации
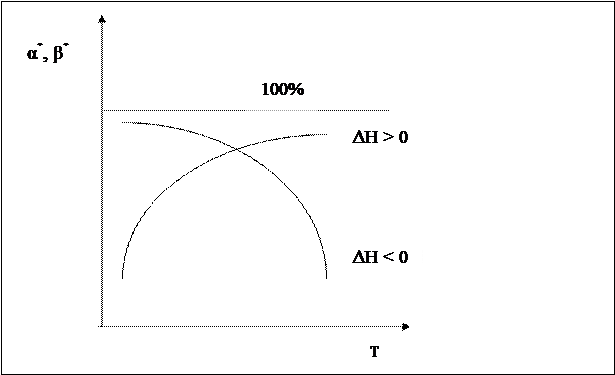
Если реакция взаимодействия А + В будет эндотермической, то получим такую же диаграмму, отличающуюся лишь тем, что энергетический уровень системы А + В ниже, чем уровень энергии системы C + D. Энергия активация прямой реакции будет выше энергии активации обратной реакции Таким образом, энергия активации эндотермического процесса всегда больше энергии активации обратного экзотермического процесса на величину теплового эффекта реакции. Следовательно, повышение температуры увеличивает в большей степени скорость эндотермической реакции. Если реакция образования целевого продукта С происходит с выделением тепла, повышение температуры увеличит в большей степени константу скорости обратной реакции, чем константу скорости прямой реакции; константа равновесия уменьшится, равновесие сместится в сторону образования исходных веществ А и В. Если ∆ Н < 0, при Т↑ Если прямая реакция эндотермическая, повышение температуры в большей степени увеличит константу прямой реакции; константа равновесия увеличится, равновесие сместится в сторону образования целевого продукта. Если ∆ Н > 0, при Т↑ Смещение равновесия означает изменение величины равновесной конверсии и выхода; в случае простого процесса они синхронно уменьшаются при увеличении температуры в экзотермических процессах и увеличиваются в эндотермических.
Степень смещения равновесия при изменении температуры зависит от величины теплового эффекта. Чем больше ∆ Н, то есть чем больше разница энергий активации прямой и обратной реакции, тем больше смещается равновесие. Если ∆ Н ≈ 0, температура почти не влияет на положение равновесия. Задача управления обратимым процессом заключается в создании условий, при которых равновесие смещено в сторону образования целевого продукта с одновременным обеспечением достаточно высокой скорости достижения равновесия. Пусть протекает простая необратимая реакция типа
Из уравнения видно, что для некоторой постоянной конверсии α А скорость процесса при повышении температуры должна возрастать за счет увеличения члена При Т↑ если Кс ↓ r↓, если Кс ↑ r↑ Первый случай реализуется в экзотермических реакциях. В обратимых экзотермических реакциях при повышении температуры скорость сначала возрастает за счет увеличения
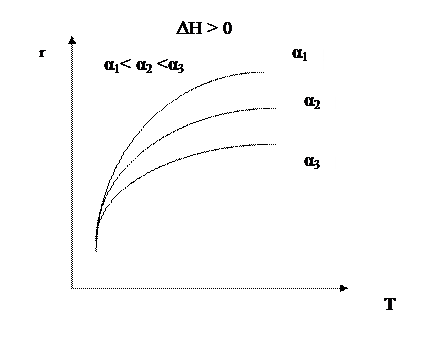
В области низких температур при повышении температуры скорость процесса увеличивается за счет увеличения скорости прямой экзотермической реакции; обратная эндотермическая реакция при этих температурах еще практически не протекает. При переходе к более высоким температурам начинает протекать обратная реакция, ее скорость в большей степени возрастает под воздействием температуры, чем скорость экзотермической реакции. В результате суммарная скорость процесса начинает уменьшаться. Температура, при которой достигается максимальная скорость, называется оптимальной. Из уравнения также видно, что при увеличении конверсии α А скорость процесса уменьшается.
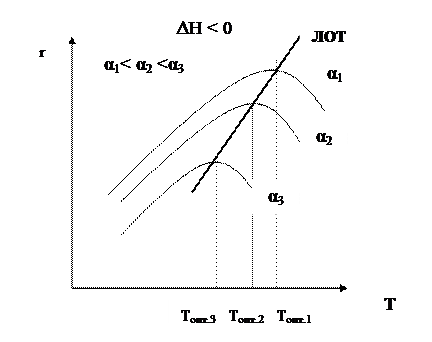
Если α 2 > α 1, кривая зависимости r =f (T) располагается ниже кривой, соответствующей α 1. Если α 3 > α 2, то соответствующая кривая располагается еще ниже и т.д. Это связано с тем, что для того, чтобы достичь более высоких значений конверсии, требуется больше времени (максимальная конверсия достигается в момент наступления равновесия). Но по мере приближения к равновесию скорость процесса падает и в момент равновесия она равна нулю. Для каждой величины конверсии получается своя кривая зависимости r =f (T), причем, чем выше конверсия, тем ниже расположена эта кривая и тем меньше оптимальная температура. Линия, соединяющая максимумы полученных таким образом кривых, является линией оптимальных температур (ЛОТ). Эта линия показывает, как нужно изменять температуру в ходе процесса для того, чтобы увеличить степень превращения реагентов, работая все время при максимально возможных в этих условиях скоростях процесса. В простых обратимых эндотермических процессах скорость процесса при повышении температуры возрастает и за счет увеличения члена
Установим зависимость конверсии от температуры α = f (T) для тех же реакций. Для обратимого экзотермического процесса зависимость α = f (T) при τ = const сначала увеличивается, достигая максимального значения, а затем снижается. Снижение α А связано с достижением равновесной конверсии, которая при повышении температуры в экзотермическом процессе уменьшается за счет смещения равновесия.
Кривая, соответствующая τ 2 > τ 1, располагается выше, чем τ 3 > τ 2 – еще выше и т.д. Кривая, соединяющая максимумы, является линией оптимальных температур (ЛОТ).
Таким образом, для обратимого экзотермического процесса не существует единой оптимальной температуры. В начале процесса нужно задавать более высокую температуру для поддержания высокой скорости процесса, а затем температуру следует снижать по ЛОТ, чтобы при достижении более высоких значений конверсии поддерживать максимально возможную скорость процесса. Для простого обратимого эндотермического процесса вопрос о влиянии температуры решается однозначно: при повышении температуры увеличивается и равновесная и фактическая степень превращения. Однако, и в этом случае есть некоторая оптимальная температура, выше которой процесс вести нерационально, поскольку конверсия увеличивается незначительно.
Для всех кривых зависимости α = f(Т) характер изменения конверсии при невысоких температурах одинаков, поскольку в этих условиях влияние термодинамических факторов (т.е. обратной реакции) незначительно. С повышением температуры влияние термодинамических факторов увеличивается. Степень этого влияния зависит от типа реакции. Для необратимых реакций кривые зависимости α = f(Т) асимптотически приближаются к единице. Для обратимых реакций они ограничиваются кривой равновесной степени превращения, которая для экзотермических процессов с повышением температуры уменьшается, а для эндотермических процессов возрастает. 2) Влияние концентрации участников реакции Суммарная скорость обратимого процесса равна
При увеличении начальной концентрации исходных реагентов А и В увеличиваются их текущие концентрации, а, следовательно, скорость прямой реакции и скорость всего процесса в целом (химическое равновесие устанавливается быстрее). Константа равновесия Н2 + I2 ↔ 2HI; Пусть С0 (I2) = 1 моль/л; С0 (Н2) = 1 моль/л. К моменту установления равновесия образовалось х моль HI, при этом в реакцию вступило х/2 моль I2 и Н2. Равновесные концентрации I2 и Н2 равны (1 – 0, 5х).
Состав равновесной смеси: 0, 7 моль/л HI, (1-0, 7/2) =0, 65 моль/л Н2 и I2. Равновесная конверсия водорода равна Увеличим начальную концентрацию йода: С0 (I2) = 2 моль/л; С0 (Н2) = 1 моль/л. Равновесная смесь содержит х моль HI, (2-0, 5) моль I2 и (1-0, 5х) моль Н2.
Состав равновесной смеси: 0, 9 моль/л HI, (2-0, 9/2) = 1, 55 моль/л I2; (1-0, 9/2) = 0, 55 моль/л Н2. Равновесная конверсия водорода равна При увеличении концентрации одного из реагентов увеличивается равновесная конверсия другого реагента, выход целевого продукта увеличивается. Таким образом, в случае обратимого процесса также целесообразно работать с высококонцентрированным сырьем и можно использовать избыток одного из реагентов. Это позволяет не только поддерживать высокую скорость процесса в течение необходимого времени, но и сдвигает равновесие в сторону образования целевого продукта. Равновесие обратимого процесса можно сместить вправо также уменьшая концентрацию продуктов С и D, выводя их из реакционной зоны. Константа равновесия Кс при этом также не изменяется: во сколько раз уменьшается произведение концентраций продуктов, во столько же раз уменьшается и равновесная концентрация исходных реагентов. При заданных значениях начальных концентраций реагентов это означает увеличение степени переработки сырья. Уменьшение концентрации продуктов в реакционной смеси приводит к уменьшению скорости обратной реакции и к увеличению суммарной скорости процесса. Способы удаления продуктов из зоны реакции могут быть разные. Продукт либо химически связывают вводимым извне веществом, например, СО + Н2О ↔ Н2 + СО2 СО2 + СаО → СаСО3↓, либо переводят в другое фазовое состояние (конденсируют или кристаллизуют при охлаждении, испаряют или возгоняют при нагревании и т.д.). В последнем случае, как правило, используют циркуляционную схему процесса. 3) Влияние давления Скорость обратимого газофазного процесса
зависит от давления, причем тем больше, чем больше отличаются порядки прямой и обратной реакции. Если реакция протекает без изменения числа молей, скорость обратимого процесса не зависит от давления. Если число молей в результате реакции уменьшается, увеличение давления приводит к увеличению скорости процесса. В противном случае повышение давления вызывает уменьшение скорости установления химического равновесия. при р↑ r = const, если r↑, если r↓, если Давление также влияет на положение равновесия. Для анализа этого влияния воспользуемся уравнением Константа равновесия КР зависит только от температуры, поэтому при Т = const, КР = const. Если Если Если Таким образом, если число молей в результате реакции уменьшается, увеличение давления способствует сдвигу равновесия в сторону образования целевого продукта и увеличению скорости процесса. Если число молей в результате прямой реакции увеличивается, для увеличения скорости процесса и сдвига равновесия в сторону образования целевого продукта нужно уменьшить давление в системе. Если Обобщая вышесказанное, можно сделать выводы, что 1) для эффективного проведения простого обратимого гомогенного процесса необходимо не только обеспечить высокую скорость процесса, но и сместить равновесие в сторону образования целевого продукта; 2) равновесие можно смещать, используя такие технологические параметры, как температура, давление, концентрация. Выбор метода смещения определяется расчетами. 3) Для обратимого экзотермического процесса требования кинетики и термодинамики находятся в противоречии, поэтому регулирование температуры необходимо осуществлять по линии оптимальных температур. 4) Повышение давления благоприятно лишь для газофазных процессов, протекающих с уменьшением числа молей. Но и в этом случае существует некоторое оптимальное давление, выше которого работать нецелесообразно из-за экономических соображений. 5) В обратимых процессах желательно осуществлять ступенчатое удаление продуктов реакции из реакционной зоны, а также работать с избытком одного из реагентов, причем вводить его можно тоже ступенчато. 6) В случае, когда условия, благоприятствующие смещению равновесия в сторону образования целевого продукта, приводят к уменьшению скорости процесса, в начале поддерживают высокую скорость, а затем создают условия для получения максимально возможного равновесного выхода. 7) Чтобы сохранить достаточно высокую скорость обратимого процесса (производительность реактора), процесс осуществляют до степени превращения реагентов 0, 8 – 0, 9 от равновесного значения.
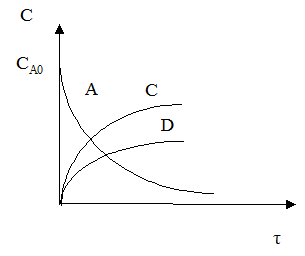
Лекция № 5 Закономерности управления сложными процессами Сложными называются процессы, в которых наряду с реакцией, приводящей к образованию целевого продукта (целевой реакцией), протекают побочные реакции. Характеристика модели: 1) Выход целевого продукта не равен конверсии реагента β ≠ α; 2) Выход целевого продукта, даже теоретически не может достигнуть значения 100% β < 100%. Объекты управления: Наряду со скоростью процесса, положением равновесия (если реакция обратима) объектом управления в сложных процессах становится селективность. r = f (С, р, t, kat) или r = f (W, ω, f) в зависимости от лимитирующей стадии α * = f (C, p, t) S = f (C, p, t, kat) Инструменты управления: кинетические параметры. Рассмотрим, как влияют основные технологические параметры на скорость и селективность сложного необратимого гомогенного процесса. Пусть протекают две параллельные реакции, в которых расходуется реагент А. Кинетические кривые для сложно-параллельного процесса имеют вид, представленный на рисунке. Концентрация исходного реагента А уменьшается во времени, а концентрация продуктов C и D увеличивается, причем скорость увеличения зависит от порядка реакции. При n1 > n2 быстрее увеличивается концентрация целевого продукта.
Для анализа воспользуемся понятием дифференциальной селективности, под которой понимают отношение скорости целевой реакции (скорости образования целевого продукта) к общей скорости процесса (скорости расходования реагента).
При условии, что
Используя полученную формулу, проведем анализ влияния начальной концентрация реагента, степени его превращения, давления и температуры на селективность процесса. Характер влияния концентрации, давления и конверсии зависит от соотношения порядков целевой и побочной реакции. Если порядки равны, начальная концентрация, давление и конверсия не влияют на селективность процесса. Если порядок целевой реакции выше, селективность увеличивается при повышении концентрации реагента и давления и уменьшается при достижении высоких конверсий. Если порядок целевой реакции ниже, чем порядок побочной реакции, при увеличении концентрации реагента и давления селективность должна понижаться, а увеличение времени реакции (конверсии реагента) приводит к повышению селективности. При СА0 ↑ rхим. р. ↑ если n1 = n2 s = const если n1 > n2 s↑ если n1 < n2 s↓
При α А↑ rхим. р.↓ если n1 = n2 s = const если n1 > n2 s↓ если n1 < n2 s↑ Для газофазных реакций
При р↑ rхим. р. ↑ если n1 = n2 s = const если n1 > n2 s↑ если n1 < n2 s↓
Влияние температуры зависит от соотношения энергий активации целевой и побочной реакции. При t ↑ rхим. р. ↑ если Еа1 = Еа2 s = const если Еа1 > Еа2 s↑ если Еа1 < Еа2 s↓ Рассмотрим другой тип сложных процессов – сложно-последовательный необратимый гомогенный процесс (консекутивный).
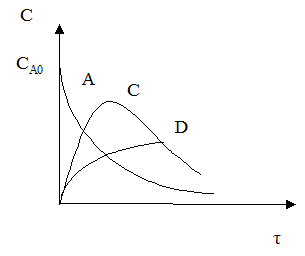
С течением времени концентрация реагента А снижается. Концентрация промежуточного продукта С вначале увеличивается, достигает своего максимального значения, затем убывает. Концентрация продукта D увеличивается по мере протекания реакции. Величина максимальной концентрации продукта С зависит от соотношения скоростей каждой из последовательных стадий.
Если целевым продуктом является продукт С, то селективность процесса равна
При увеличении начальной концентрации реагента СА0 ↑ rхим. р. ↑ если n1 = n2 s = const если n1 > n2 s↑ если n1 < n2 s↓ При увеличении степени превращения реагента α А↑ rхим. р, s↓ При увеличении температуры t ↑ rхим. р. ↑ если Еа1 = Еа2 s = const если Еа1 > Еа2 s↑ если Еа1 < Еа2 s↓ На селективность газофазных процессов будет оказывать влияние давление.
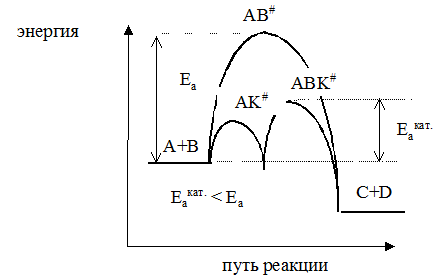
При р↑ rхим. р. ↑ если n1 = n2 s = const если n1 > n2 s↑ если n1 < n2 s↓ Анализ показал, что в некоторых случаях изменение технологического параметра может привести к увеличению скорости процесса, а селективность при этом уменьшится или к увеличению селективности при снижении скорости процесса. Выбор оптимального значения этого технологического параметра производится путем технологических и экономических расчетов. Очень часто предпочитают получить высокую селективность, особенно если сырье имеет высокую стоимость, высока стоимость отделения и очистки целевого продукта от побочных продуктов, а скорость процесса невелика. Обобщая вышесказанное, можно сделать следующие выводы: 1) При разработке технологии сложных процессов основное внимание следует уделить селективности процесса. Другими словами, нужно найти условия, обеспечивающие преимущественное протекание целевой реакции. 2) Условия проведения процесса определяются на основании экспериментального изучения кинетики процесса (определения порядка всех реакций, энергии активации и др.). Найденные условия можно менять в узком диапазоне. 3) Самое лучшее решение в задаче повышения селективности процесса – это нахождение селективного катализатора. Лекция № 6 Закономерности управления каталитическими процессами Катализ – это явление изменения скорости химической реакции под воздействием малых количеств веществ – катализаторов, которые, участвуя в процессе, восстанавливают свой состав в конце каталитического цикла. Катализ является наиболее эффективным и рациональным средством регулирования скорости химической реакции. Более 80% промышленных процессов являются каталитическими. Катализатор может увеличивать скорость химической реакции (положительный катализ) или уменьшать ее (отрицательный катализ). В последнем случае катализатор называют ингибитором. Торможение (ингибирование) нежелательных процессов играет в химической технологии не менее важную роль, чем каталитическое ускорение реакции. Ускоряющее действие катализаторов принципиально отличается от действия других факторов, интенсифицирующих процесс. Концентрация реагирующих веществ и давление увеличивают общее число столкновений молекул, температура, различные виды облучения увеличивают энергию сталкивающихся молекул. Катализатор снижает энергию активации процесса в результате изменения реакционного пути. Пусть протекает реакция аА + вВ → сС + dD. Чтобы вещества А и В образовали продукты С и D, они должны преодолеть некоторый энергетический барьер. На это затрачивается энергия активации Еа. Молекулы, обладающие этой избыточной энергией. Образуют неустойчивую группировку, называемую активированным комплексом АВ#. Скорость реакции непосредственно зависит от значения энергии активации; если она мала, то в единицу времени большее количество молекул преодолеют энергетический барьер, и скорость реакции будет высокой. Если энергия активации велика, то реакция идет медленно. Катализатор тем или иным способом изменяет реакционный путь. Например, он взаимодействует с молекулой А, образуя некоторый активированный комплекс АК#. Этот комплекс взаимодействует с молекулой вещества В, образуя новое неустойчивое соединение АВК#, которое, разрушаясь, дает продукты С и D и катализатор в первоначальном виде. А + К → АК# АК# + В → АВК# АВК# → С + D + К
Таким образом, процесс разбивается на ряд стадий, каждая из которых требует преодоления меньшего энергетического барьера, чем в случае некаталитической реакции. Мерой ускоряющего действия катализатора является величина относительной активности катализатора. Относительная активность рассчитывается как отношение константы скорости каталитической реакции к константе скорости некаталитической реакции.
Видно, что даже незначительное уменьшение энергии активации может увеличить скорость реакции в десятки, сотни и более раз. В качестве примера можно привести реакцию окисления сернистого ангидрида, осуществляемую при температуре 693 К в присутствии катализатора V2O5. Еа =420000 Дж/моль, Еакат. = 268000 дж/моль
Скорость окисления SO2 в присутствии катализатора V2O5 возрастает в 3 . 1011 раз. Даже, если реакция из-за своей малой скорости практически совсем не протекает. Использование подходящего катализатора приводит к полному и быстрому химическому превращению. Однако следует помнить, что: 1) Использование катализатора не может вызвать термодинамически невозможную реакцию. Если ∆ G > 40 кДж/моль, реакция термодинамически невозможна, катализатор искать бесполезно. Если 0 < ∆ G < 40 кДж/моль, и тем более, если ∆ G < 0, подходящий катализатор искать можно и нужно. 2) Катализатор не может смещать положение равновесия в обратимых процессах, так как в равной степени ускоряет и прямую и обратную реакции, способствуя более быстрому достижению равновесия. В зависимости от фазового состояния катализатора и реагентов различают катализ гомогенный и гетерогенный. Оценим преимущества и недостатки этих двух видов катализа.
Основным недостатком гомогенного катализа является сложность его выделения из реакционной среды. Часть катализатора, а иногда и весь катализатор теряется безвозвратно. Это увеличивает экономические затраты на производство, ухудшает качество продукта, увеличивает количество сточных вод и отходов. Основными проблемами при использовании гетерогенного катализатора является необходимость решения вопросов, связанных с интенсификацией массо- и теплообменных процессов. Методы управления гомогенно-каталитическими процессами мало чем отличаются от приемов интенсификации гомогенных некаталитических процессов, хотя участие катализатора в процессе вносит свою специфику. Например, известно, что согласно закону действующих масс, скорость реакции должна возрастать пропорционально концентрации реагирующих веществ. Однако в гомогенно-каталитическом процессе А → С возможен случай, когда скорость реакции, увеличиваясь по мере увеличения концентрации реагента, достигает некоторой величины и перестает изменяться. Причиной этого является то, что общая скорость процесса
лимитируется стадией разрушения промежуточного комплекса катализатора с реагентом А. А + К → АК# АК# → C + К Скорость разрушения этого комплекса зависит от его концентрации, которая, в свою очередь, зависит от концентрации катализатора. Если концентрация катализатора мала, он весь связан в комплекс, и увеличение концентрации реагента А бесполезно. Таким образом, следует помнить, что катализатор активно участвует в процессе; его концентрация может оказаться эффективным инструментом управления процессом. Основные стадии и кинетические особенности гетерогенно-каталитических процессов
В общем случае процесс гетерогенного катализа складывается из следующих стадий: 1 – внешняя диффузия молекул реагентов из ядра потока к поверхности катализатора через пограничный слой δ; 2 – внутренняя диффузия молекул в порах катализатора; 3 – активированная адсорбция молекул на поверхности катализатора с образованием поверхностных непрочных химических соединений – активированных комплексов; 4 – перегруппировка атомов с образованием поверхностных комплексов «продукт-катализатор» (химическая реакция); 5 – десорбция молекул продуктов с поверхности; 6 – внутренняя диффузия молекул продуктов в порах катализатора; 7 – внешняя диффузия молекул продуктов от поверхности катализатора в ядро потока через пограничный слой. Стадии 3, 4, 5 являются химическими, 1, 2, 6, 7 – массообменные (диффузионные). В зависимости от того, какая из этих стадий является лимитирующей, различают три области протекания гетерогенно-каталитического процесса: внешнедиффузионную, внутридиффузионную и кинетическую. Область протекания определяется экспериментально. Если скорость процесса зависит от линейной скорости потока W при постоянном отношении объема катализатора и объема реакционной смеси, процесс протекает во внешнедиффузионной области, то есть лимитируется стадией внешней диффузии веществ через пограничный слой. Эта область является самой неблагоприятной для проведения процесса: - работает только внешняя поверхность катализатора, которая значительно меньше внутренней; - при экзотермических реакциях катализатор работает в жестком температурном режиме, так как выделяющееся тепло не успевает отводиться с поверхности катализатора; Популярное:
|
Последнее изменение этой страницы: 2017-03-11; Просмотров: 687; Нарушение авторского права страницы
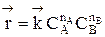 ,
, 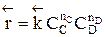 .
.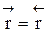 ; наступает химическое равновесие. После достижения равновесия соотношение участников реакции остается постоянным во времени. Это соотношение называется константой равновесия.
; наступает химическое равновесие. После достижения равновесия соотношение участников реакции остается постоянным во времени. Это соотношение называется константой равновесия.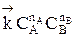 =
= 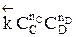
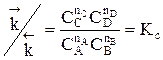
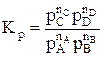
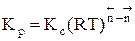 , где
, где 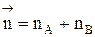 - суммарный порядок прямой реакции,
- суммарный порядок прямой реакции, 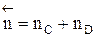 - суммарный порядок обратной реакции.
- суммарный порядок обратной реакции.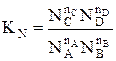 .
.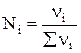 , ν i – число молей данного компонента. Тогда
, ν i – число молей данного компонента. Тогда 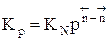 .
.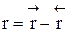 . Она уменьшается во времени и при
. Она уменьшается во времени и при 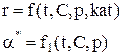
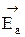 . Для того, чтобы прореагировали вещества C и D, они тоже должны обладать энергией активации
. Для того, чтобы прореагировали вещества C и D, они тоже должны обладать энергией активации  . Как видно из рисунка, энергия активации обратной реакции больше энергии активации прямой реакции
. Как видно из рисунка, энергия активации обратной реакции больше энергии активации прямой реакции 
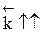 ,
, 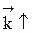
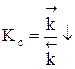
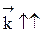 ,
, 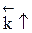 ,
, 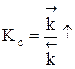 .
.
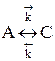 .
.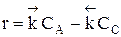
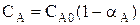
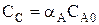
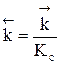
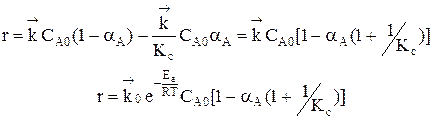
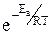 , а с другой стороны, может уменьшаться, если уменьшается константа равновесия Кс, или увеличиваться, если Кс увеличивается.
, а с другой стороны, может уменьшаться, если уменьшается константа равновесия Кс, или увеличиваться, если Кс увеличивается.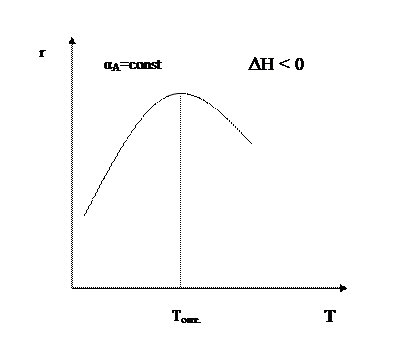
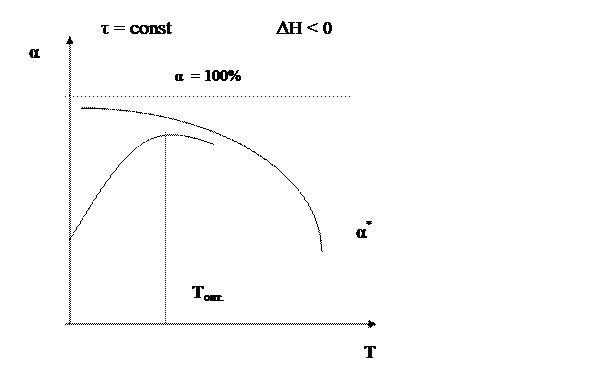
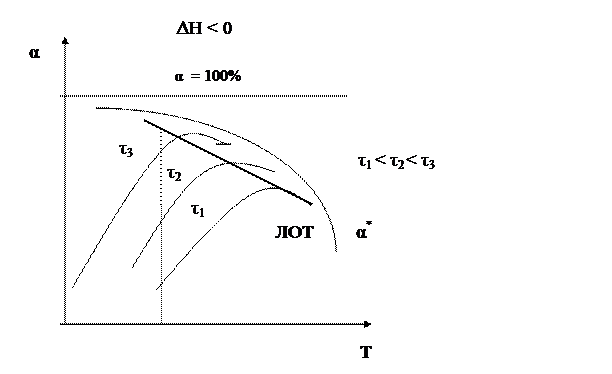
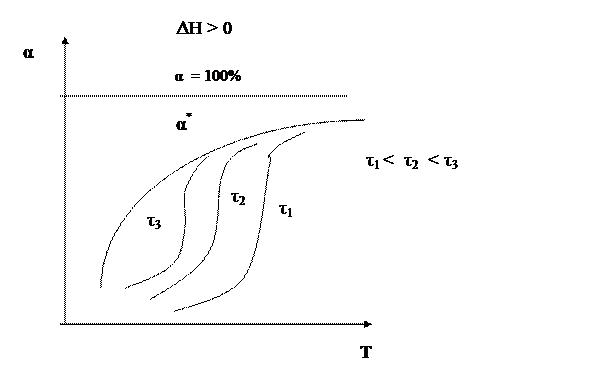
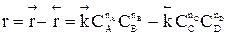
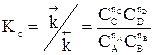 при этом не меняется: во сколько раз увеличивается произведение равновесных концентраций реагентов, во столько раз увеличивается и произведение равновесных концентраций продуктов. Однако равновесный состав смеси будет другой. Покажем это на примере образования йодоводорода.
при этом не меняется: во сколько раз увеличивается произведение равновесных концентраций реагентов, во столько раз увеличивается и произведение равновесных концентраций продуктов. Однако равновесный состав смеси будет другой. Покажем это на примере образования йодоводорода.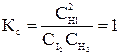
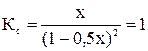 ; 0, 75 х2 + х – 1 = 0; х = 0, 7
; 0, 75 х2 + х – 1 = 0; х = 0, 7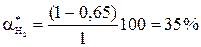 .
.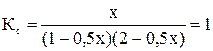 ; 0, 75х2 + 1, 5х – 2 = 0; х = 0, 9
; 0, 75х2 + 1, 5х – 2 = 0; х = 0, 9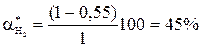 .
.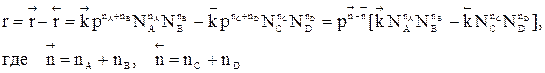
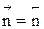 ;
; 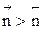
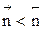
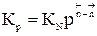 .
.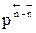 , КN должна увеличиться, равновесие сдвигается в сторону образования целевого продукта.
, КN должна увеличиться, равновесие сдвигается в сторону образования целевого продукта.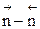 близко к нулю, использовать давление для повышения эффективности процесса нецелесообразно, так как затраты намного превысят полученный эффект.
близко к нулю, использовать давление для повышения эффективности процесса нецелесообразно, так как затраты намного превысят полученный эффект.