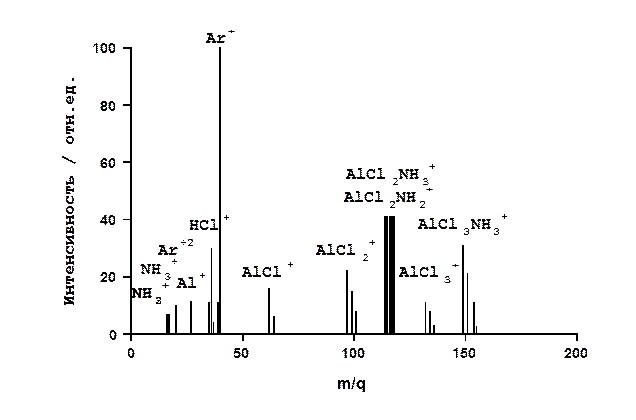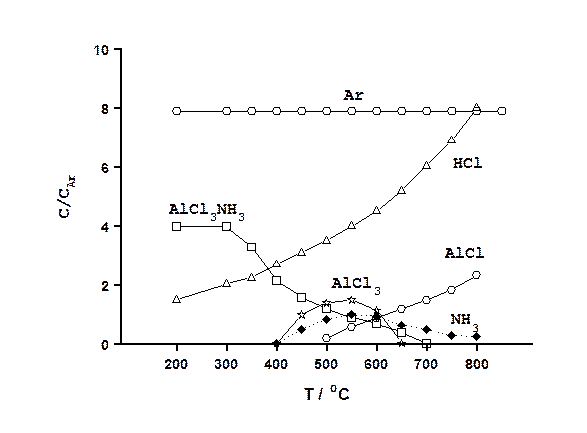|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Интерференционная голография. ⇐ ПредыдущаяСтр 9 из 9
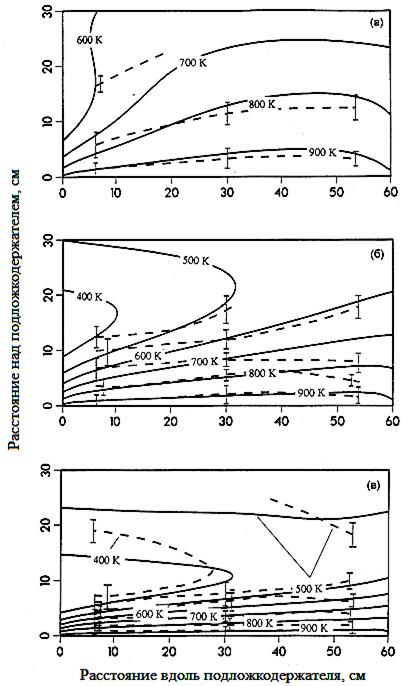
В противоположность использованию мощных лазеров, требующихся для рамановской спектроскопии, интерференционная голография может быть реализована с использованием дешевых лазеров, имеющих средние мощности в несколько милливатт. Однако в случае интерференционной голографии свет падает на движущиеся оптические компоненты системы. Кроме того, температурные колебания окружающего воздуха (либо охлаждающей воды) могут приводить к значительным ошибкам. Интерференционная голография является более широко используемым методом, так как применима для любых газов, хотя некоторое ухудшение чувствительности и точности измерений возникает при использовании газов, характеризующихся малыми значениями показателя преломления (гелий). Главным преимуществом интерференционной голографии является возможность получения информации в режиме реального времени о температурном поле в интересующей области реактора. Как уже упоминалось, интерференционная голография основана на регистрации различия в длинах оптических путей, возникающих из-за разных показателей преломления среды. В простейшем случае газа с однородной плотностью в направлении распространения лазерного луча полосы на интерференционной голограмме соответствуют изотермам температуры. В более сложном случае, когда температура газа изменяется в направлении распространения лазерного луча, требуется проведение обратных расчетов для извлечения информации о температурных полях. Используя интерференционную голографию удалось измерить температурные профили в застойной зоне над пьедесталом в вертикальном CVD реакторе. На рис. 22 представлены полученные профили температуры газа над пьедесталом.
Рис.22. Измеренные (пунктир) и рассчитанные (сплошные линии) изотермы над подложкодержателем в горизонтальном реакторе для различных газов-носителей и их расходов. а – 2 л/мин., водород; б - 8 л/мин., водород; в - 2 л/мин., азот; Обращает на себя внимание наличие «холодного пальца» в случаях (б) и (в). Определение состава реакционной газовой фазы в зоне осаждения.
Пространственное распределение компонентов реакционной газовой среды в CVD реакторе, определяемое скоростями протекания гомогенных и гетерогенных химических реакций, массо- и теплопереноса, относится к важнейшим характеристикам процесса осаждения и по существу является отражением его механизма и реакционной схемы. Именно, поэтому исследованию состава реакционной газовой фазы, образующейся в зоне осаждения при различных условиях протекания процесса, уделяется большое внимание. Информация о природе химических соединений, присутствующих в реакционной газовой фазе, их концентрации и влиянии на нее технологических параметров процесса, является необходимой для понимания механизма CVD процесса. Контроль за расходованием реагентов, при протекании газовой смеси через реактор, позволяет оценить суммарную эффективность процесса осаждения. Выяснение механизма газофазных реакций позволяет идентифицировать те химические компоненты, которые поступают на поверхность подложки и определяют гетерогенные реакции, приводящие к образованию слоя осаждаемого материала. Это особенно важно для понимания влияния условий осаждения слоев на их состав и физико-химические свойства. Кроме того, очевидно, что построение модели процесса осаждения невозможно без наличия информации о составе реакционной газовой фазы. Идеальный метод изучения состава реакционной газовой среды должен обеспечивать минимальное воздействие на нее при отборе пробы для анализа, возможность определения отдельных компонентов смеси, быть чувствительным, количественным и обеспечивать высокое разрешение в условиях быстро изменяющихся концентрационных и температурных полей. В связи с тем, что ни один из известных методов анализа газовой среды не отвечает всем перечисленным требованиям, только использование их комбинаций позволяет получать достоверные сведения о составе газовой фазы.
Масс-спектрометрия
Масс-спектрометрия – хорошо разработанный аналитический метод, в котором анализируемые газы сначала ионизируются, а затем пучок заряженных частиц разделяется с помощью электрического и магнитного поля на фракции с одинаковым отношением массы к заряду (m/e) и определяется масса иона. Этот метод реализуется в специальных приборах, называемых масс-спектрометры. К настоящему времени разработаны несколько типов масс-спектрометров. Рассмотрим простую блок-схему, общую для всех масс-спектрометров. Основной узел масс-спектрометра - масс-анализатор, где под действием электрического и магнитного поля происходит процесс разделения пучка ионов на фракции с одинаковым отношением массы к заряду (m/e). Поскольку анализировать можно только заряженные пучки, а в большинстве случаев нужно исследовать нейтральные частицы, необходимо превратить их в ионы. Для этого служит ионный источник, в котором нейтральные частицы подвергаются ионизации. Существует большое количество методов ионизации, среди которых наиболее широко используется электронный удар. Наконец, третья обязательная деталь - регистрирующее устройство, с помощью которого можно определить количество ионов с данным m/e. Это могут быть фотопластина (масс-спектрограф), электрометр или электронный умножитель (масс-спектрометр). В современном приборе регистрирующее устройство непосредственно связано с компьютером, который производит обработку результатов и управляет экспериментом. Масс-спектрометр - вакуумный прибор, снабженный специальной системой откачки. В масс-анализаторе заряженные частицы должны проходить расстояние в несколько метров, не сталкиваясь с молекулами остаточных газов. Для этого требуется вакуум ≤ 10-5 Па. На лучших моделях современных приборов достигается разрежение 10-7 Па. Масс-анализаторы принято делить на статические и динамические. Статические приборы используют постоянное, а динамические - переменное электрическое и магнитное поле. Основные принципы работы масс-спектрометра и его характеристики удобно проиллюстрировать на простейшем масс-спектрометре с магнитным анализатором секторного типа (рис.23). Пучок ионов из ионизационной камеры проходит постоянную разность потенциалов U = 3-5 кВ и попадает в однородное магнитное поле напряженности H. Здесь на него действует сила Лоренца F = e[H.υ ], где e - заряд частицы, υ - ее скорость. Все ионы одинакового заряда приобретают одинаковую кинетическую энергию Екин = U.е, и их скорость связана с массой известным соотношением υ = (2U.е/m)1/2. Сила Лоренца заставляет ион отклоняться от прямолинейного движения и выводит его на круговую орбиту радиуса r: e.υ H = m .υ 2 /r. Подставляя значение υ , получаем r = 1/ H(2m .U/e)1/2.
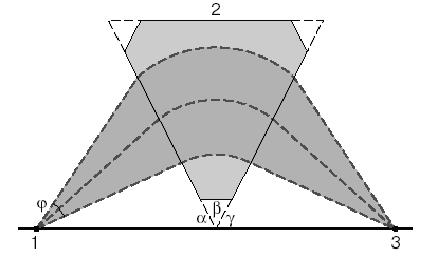
Рис.23. Фокусирующее действие секторного поля. 1- источник ионов; 2 – секторное магнитное поле; 3 – детектор; φ – угол расходимости ионного пучка с одинаковым массовым числом М.
Таким образом, частицы с различными m/e по-разному отклонятся в магнитном поле и окажутся в разных точках детектора. Если детектор - фотопластина, мы получим на фотографии масс-спектр пучка ионов. В большинстве масс-спектрометров используют другой прием: радиус r оставляют постоянным, а сканирование по массам производят изменяя H. Фокусирующее действие секторного магнитного поля проиллюстрировано на рис.23. Как видно из рис.23, фокусировка осуществляется по углу, то есть ионы, имеющие одинаковое значение m/e, но входящие под разными углами в магнитное поле, после прохождения этого поля снова собираются в одну точку. На практике происходит некоторое уширение изображения, и последнее получило название " сферическая аберрация". Хроматическая аберрация связана с разбросом ионов по энергии. Приборы, в которых осуществляется фокусировка ионов как по углу, так и по энергии (скорости), называются приборами с двойной фокусировкой. Эти приборы по техническим характеристикам существенно превосходят приборы с одной фокусировкой. Основными характеристиками масс-спектрометров являются диапазон измеряемых масс и разрешающая способность (разрешение). Разрешающая способность 1000 означает, что сигналы, отвечающие m/e, равные 1000 и 1001, будут фиксироваться отдельно на регистрирующем устройстве, и их перекрывание не будет превышать 10% от полусуммы их интенсивностей (уровень фона). В некоторых случаях разрешение указывается для 50% уровня фона. В наиболее распространенных и относительно дешевых статических приборах диапазон измеряемых масс лежит в интервале 500-1500 атомных единиц массы, а разрешение - в интервале 200-800. В последние десятилетия широкое применение нашли времяпролетные масс-спектрометры и приборы ион-циклотронного резонанса. Во времяпролетном масс-спектрометре пучок ионов, пройдя ускоряющую разность потенциалов (несколько киловольт), летит в бесполевом пространстве к детектору. Под действием одинакового ускоряющего напряжения ионы с разным m/e приобретают разную скорость и регистрируются в разное время. Таким образом происходит разделение по отношению массы к заряду. Времяпролетные масс-спектрометры работают в импульсном режиме, длительность импульса около 10 -4 с. Они незаменимы при исследовании процессов, быстро протекающих во времени. В лучших образцах диапазон измеряемых масс и разрешающая способность достигают нескольких тысяч. Приборы ион-циклотронного резонанса имеют рекордно высокую разрешающую способность, достигающую нескольких миллионов. В этих приборах ионы дрейфуют из ионного источника в резонансную полость под действием постоянного магнитного H и электрического E полей, направленных перпендикулярно друг к другу. Траектория движения - циклоида, являющаяся результатом равномерного прямолинейного движения и движения по окружности с циклотронной частотой (угловой скоростью) w = eH/m. В резонансной полости ионный пучок попадает под действие поляризованного радиочастотного поля частоты n. При равенстве w/2p=n наступает резонанс, происходит поглощение энергии поля ионами. При постоянной n, меняя H, можно достичь резонанса последовательно по различным m/e. Из изложенного легко видеть, что современные масс-спектрометры позволяют определять массы тяжелых молекул вплоть до сотен а.е.м. с точностью до одной массовой единицы. Основным преимуществом масс-спектрометрии для исследования CVD процессов является ее универсальность (любые системы реагентов могут быть исследованы, так как все молекулы имеют масс-спектры) и возможность проводить количественные измерения. С помощью масс-спектрометрии можно получать точные значения относительных количеств различных компонентов исследуемого газа и при благоприятных условиях определять абсолютные парциальные давления реагентов. Главным недостатком применения масс-спеткрометрии для исследования CVD процессов является необходимость осуществления отбора пробы анализируемой реакционной газовой фазы. В этой связи, практическая реализация масс-спектрометрии для диагностики процессов химического осаждения из газовой фазы связана с рядом трудностей, в первую очередь относящихся к способу отбора пробы анализируемой реакционной смеси. Причина трудностей состоит в том, что обычно газофазные процессы осаждения осуществляются при атмосферном давлении либо при пониженном, находящимся в интервале 100-1500 Па, а давление в анализаторе масс-спектрометра, как правило, не превышает 10-3 Па. В литературе приведены результаты исследования газофазных процессов осаждения с применением масс-спектрометрии, в которых такой перепад давления в процессе отбора пробы обеспечивался с помощью капилляров (например, рис.24). Однако, такой способ не является корректным применительно к решаемой задаче, так как в процессе перемещения пробы по капилляру ее состав может претерпевать существенные изменения. Кроме того, введение капилляра в реакционный газовый поток вызывает искажения его структуры и приводит к отклонению состава исследуемой среды. Очевидно, что абсолютно идеальное, то есть обеспечивающее отсутствие каких-либо внутренних поверхностей переходного устройства, сконструировать невозможно. Наилучший способ соединения камеры анализатора масс-спектрометра с реактором требует минимизации этой поверхности, что достигается за счет применения тонкой (толщиной 0.2-1 мм) диафрагмы с отверстием 20-60 мкм.
Рис. 24 Схема CVD реактора для исследования состава реакционной газовой фазы, образующейся при осаждении нитрида галлия.
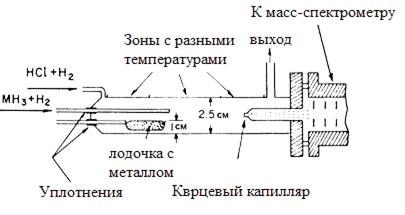
На рис. 25 приведена схема модельного реактора, разработанного на кафедре ТМЭТ СПбГПУ, для изучения состава реакционной газовой смеси, образующейся в зоне осаждения в ходе ХОГФ, и узла сочленения его с модернизированным масс-спектрометром МСХ-6. Отбор пробы из реактора в область ионизатора 1 масс-спектрометра осуществляется через тонкую кварцевую диафрагму 2, диаметр отверстия в которой составлял ~ 45 мкм, что обеспечивало давление в анализаторе 10-3 Па при рабочем давлении в реакторе 250-300 Па. Расстояние от диафрагмы до ближайшей сетки источника ионов составляет 20 мм. Диафрагма находилась при той же температуре, при которой проводился процесс химического осаждения из газовой фазы. Распределение температуры вдоль реактора регулировалось варьированием токов в многосекционнном резистивном нагревателе 3 и контролировалось термопарой 6, перемещаемой вдоль кварцевого канала. Во избежание перегрева корпуса фланец масс-спектрометра и модельный реактор снабжены водяной “рубашкой”.
Рис. 25. Схема модельного реактора для масс-спектрометрической диагностики состава газовой фазы, образующейся при газофазном осаждении.
В качестве примера рассмотрим результаты использования этой экспериментальной аппаратуры и методики исследований для изучения химического осаждения из газовой фазы при пониженном давлении пленок нитрида алюминия. Процесс основан на пиролизе паров комплекса AlCl3.NH3, помещаемого в испаритель 5. Пары комплекса из испарителя, находящегося при температуре 200-250 оС, переносились потоком аргона марки ВЧ в зону реакции. Общее давление в реакторе составляло 275 Па. При обработке масс-спектров допускали, что отношение интенсивностей молекулярного (материнского) и осколочного пиков остается постоянным при неизменной энергии ионизирующих электронов. На рис.26 приведен характерный масс-спектр газовой фазы, содержащий в основном парообразный AlCl3.NH3, полученный при температуре в реакционной зоне 300 оС. Как видно, помимо молекулярного иона AlCl3.NH3+ в спектре присутствуют также осколочные ионы NH2+, NH3+, Cl+, HCl+, AlCl+, AlCl2+, AlCl3+, AlCl2.NH2+(AlCl2.NH3+), Al+.
Рис. 26. Типичный масс-спектр газовой фазы при температуре в реакционной зоне - 300 оС.
К сожалению, низкая разрешающая способность и чувствительность используемого времяпролетного масс-спектрометра не позволили надежно идентифицировать другие осколочные ионы. Однако и полученная информация оказалась ценной с точки зрения понимания химизма процесса осаждения. Соотношение интенсивностей осколочных пиков AlCl2.NH2+(AlCl2.NH3+) и AlCl3+ свидетельствует о том, что связь Al-N в молекуле AlCl3.NH3 является более прочной чем связь Al-Cl. Это подтверждает предложенную ранее гипотезу о том, процесс пиролиза AlCl3.NH3 может протекать и без разрыва связи Al-N [12]. Однако, как показывают полученные результаты (см. рис. 27), такой механизм возможен лишь при температурах осаждения, не превышающих 700 оС, то есть когда комплекс еще не диссоциировал на трихлорид и аммиак.
Рис.27. Качественное изменение состава газовой фазы от температуры зоны пиролиза.
Как следует из приведенных на рис. 27 данных, исходный комплекс остается устойчивым в исследуемых условиях до температуры 350 оС. При более высоких температурах он частично диссоциирует, вероятнее всего по реакции AlCl3.NH3 Û AlCl3 + NH3 (4.1) Интересно отметить, что трихлорид алюминия исчезает из газовой фазы при той же температуре (650-700 оС), что и исходный комплекс. По-видимому, протекание реакции (4.1) является единственным источником его образования. Экспериментально установлено, что при температурах выше 450 oC происходит пиролиз исходного комплекса с образованием нитрида алюминия в соответствии с реакцией: AlCl3.NH3 Þ AlN + 3HCl3 (4.2) В узком интервале температур 350-450 оC, когда реакция (4.2) невозможна при исследуемых условиях, наблюдается выделение хлороводорода (см. рис.27), которое, наиболее вероятно, обусловлено образованием амидохлорида алюминия AlCl2.NH2. Несмотря на то, что из-за недостаточной разрешающей способности МСХ-6 присутствие этого соединения не обнаружено, в некоторых спектрах, зарегистрированных в этом узком интервале температур, наблюдалось аномальное соотношение интенсивностей молекулярного AlCl3.NH3+ и осколочного пиков AlCl2.NH2+(AlCl2.NH3+) - противоположное обычно наблюдаемому в спектре исходного комплекса, что может быть обусловлено только появлением нового соединения. Экспериментально было установлено, что в отличие от CVD процесса осаждения пленок GaN, основанного на пиролизе моноаммиаката хлорида галлия, травление пленок AlN газообразным хлороводородом, сопровождающееся выделением AlCl, в исследованном диапазоне температур не происходит. Вероятно монохлорид алюминия, являющийся преобладающим алюминийсодержащим компонентом реакционной среды при температурах пиролиза более 700 оС, образуется за счет восстановления водородом или диссоциации высших хлоридов. Факт протекания подобных реакций экспериментально установлен и в системе с участием галлия, однако их вклад в накопление GaCl оказался пренебрежимо малым. Вполне возможно, что при таких высоких температурах образование пленок нитрида алюминия происходит путем прямого взаимодействия монохлорида алюминия с аммиаком. Суммируя полученные результаты по исследованию состава реакционной газовой смеси, образующейся в зоне осаждения в ходе процесса ХОГФ пленок нитрида методом масс-спектроскопии можно констатировать определенное сходство в химизме процессов, протекающих при пиролизе аммиачно-хлоридных комплексов алюминия и галлия в области низких температур (до 650-700 оС). Однако, при более высоких температурах поведение систем различно. Следует отметить, что масс-спектрометр работает только с заряженными частицами, которые могут образовываться из нейтральных молекул или атомов, присутствующих в реакционной среде, различными путями, в связи с чем расшифровка спектров, то есть определение состава «материнских» молекул, особенно многоатомных, представляет собой не простую задачу, как могло показаться.
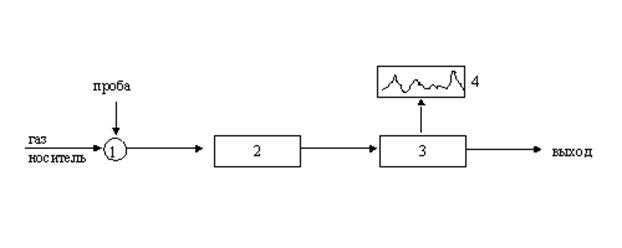
Газовая хроматография Газовая хроматография представляет собой хорошо поставленный метод разделения и идентификации различных газообразных веществ и широко используется в аналитической химии как рутинный метод анализа. В случае газовой хроматографии подвижной фазой служит газ (пар), а в зависимости от агрегатного состояния неподвижной фазы различают газоадсорбционную хроматографию (неподвижная фаза – твердое тело) и газожидкостную (неподвижная фаза – жидкость, нанесенная тонким слоем на твердый носитель). Разделение компонентов в этом методе основано на различии скоростей движения и размывания концентрационных зон исследуемых веществ, движущихся в потоке газовой фазы относительно слоя неподвижной, причем эти вещества распределяются между обеими фазами. Газ-носитель (N2, Ar, He и др.) должен обычно иметь небольшую вязкость и обеспечивать высокую чувствительность детектирования. Газохроматографическое разделение и анализ осуществляется в специальном приборе – газовом хроматографе. Общая схема хроматографа приведена на рис.28.
Рис.28. Принципиальная схема газового хроматографа: Поток газа-носителя из баллона непрерывно подается в хроматографическую колонку, а оттуда в детектирующее устройство. Оно непрерывно измеряет концентрацию компонентов у выхода из колонки и преобразует ее в электрический сигнал, регистрируемый потенциометром. В газовой хроматографии практически используются только дифференциальные детекторы (катарометры, пламенно-ионизационный, электронно-захватный и пламенно-фотометрический). На ленте самописца получается выходная кривая, которую называют хроматограммой. Полученная хроматограмма обычно представляет собой ряд пиков. Площадь пика пропорциональна количеству каждого компонента, а время выхода пика при постоянном режиме работы прибора (постоянная температура колонки и скорость газа-носителя) характеризует природу компонента. Таким образом, на одном приборе можно проводить не только разделение, но и качественный и количественный анализ смесей. Хроматографическая колонка представляет собой стеклянную или металлическую трубку. Для аналитического разделения используют насадочные колонки длиной 0.5 – 5 м и диаметром 0.2 – 0.6 см, а также капиллярные полые колонки длиной до 100 м и диаметром 0.1 – 1 мм, и капиллярные насадочные колонки длиной до 20 м. Она находится в термостате, который позволяет поддерживать температуру колонки постоянной (изотермический режим) или изменять ее во времени по заданной программе. При газоадсорбционном варианте хроматографическую колонку заполняют частицами сорбента размером 0, 1-0, 3 мм с высокоразвитой поверхностью 10-600 м2/г и с достаточной механической прочностью. В качестве адсорбентов используют оксид алюминия, активированные угли, графитированные сажи, молекулярные сита (цеолиты) или пористые полимерные сорбенты и др. Метод газовой хроматографии служит для разделения летучих веществ, к которым обычно относятся вещества с молекулярной массой приблизительно до 300, и термически стойких соединений. В газожидкостном варианте в качестве сорбента используют более сложную композицию, состоящую из твердого носителя, покрытого нелетучей в условиях проведения опыта жидкостью толщиной несколько микрон (неподвижная жидкая фаза). Механизм разделения смеси в данном случае основан на различной растворимости ее компонентов в поглощающей среде. В качестве неподвижных жидких фаз используются практически все основные классы органических соединений как низко, так и высококипящие, в частности углеводороды, амиды, простые и сложные эфиры, нитрилы, кислоты, полигликоли, полиэфиры, силиконовые жидкости с различными функциональными группами и т.д. Методы газовой хроматографии очень чувствительны. Для проведения газовой хроматографии часто вполне достаточно нескольких кубических миллиметров газа, долей микролитра жидкости или долей микрограмма твердого вещества. Анализируемая проба вводится в поток газа при повышенной температуре дозатором (например, микрошприцем) через тонкую мембрану из эластичного материала. Существую также автоматические системы ввода проб. Преимущество газовой хроматографии состоит в ее универсальности – большинство стабильных молекул может быть определено этим методом, причем концентрация компонентов, присутствующих в газовой смеси даже в следовых количествах, могут быть надежно измерены. Главными недостатками использования газовой хроматографии для исследования состава реакционной газовой фазы, образующейся в CVD реакторе, обусловлены необходимостью отбора пробы, поэтому этот метод не позволяет проводить анализ в режиме реального времени. Химически активные компоненты смеси не могут непосредственно быть обнаруженными, так как они подвергаются превращениям в ходе анализа. Кроме того, иногда возникают проблемы с определением компонентов, присутствующих в малых концентрациях (что характерно, для реагентов, используемых в CVD процессах). Многие исследователи отмечают высокую трудоемкость метода, связанную с подбором оптимального состава неподвижной фазы и поиском наиболее подходящих режимов анализа. Именно по этим причинам этот метод использовался для исследования состава реакционной смеси в CVD процессах только в нескольких случаях. В частности газовая хроматография использовалась для изучения процесса образования пленок нитрида кремния в системе реагентов SiH4-NH3. Схематическое изображение экспериментальной установки показано на рис. 29. Авторы этих исследований контролировали с помощью хроматографии SiH4, H2, N2, NH3 и Ar и опубликовали концентрационные профили этих компонентов по высоте над подложкодержателем. Концентрация водорода плавно уменьшается с увеличением расстояния от поверхности подложки, тогда как концентрация силана увеличивается. Обнаруженное изменение концентрации молекулярного азота вызвало наибольший интерес (рис. 30), хотя какие-либо понятные объяснения выявленной картине предложить не удалось.
Рис. 29. Схема установки для исследования состава газовой фазы методом хроматографии
Рис.30. Распределение концентрации азота над поверхностью подложки при осаждении нитрида кремния. Газовая хроматография была успешно применена для исследования процесса осаждения углерода из метана. Авторы с помощью этого метода анализа контролировали процесс образования H2 и различных алифатических и ароматических углеводородов в течение CVD процесса. Было установлено присутствие в реакционной зоне следующих молекул С2H6, С2H4, С2H2, С6H6, С3H6, С3H8 и С4Hn. Кроме того, авторы этой же работы применяли хроматографию для оценки герметичности реактора по присутствию СО2. Сравнительно недавно хроматография использовалась для исследования состава газовой смеси углеводородов с водородом, образующейся при осаждении алмазоподобных пленок. Кроме того, имеются сведения об использовании хроматографии для исследования процессов осаждения кремния из хлорсиланов и осаждения селенида цинка из диметил цинка. В целом, из приведенных примеров следует, что газовую хроматографию целесообразно применять для исследования CVD процессов только в том случае, когда вероятно присутствие в газовой фазе высоко стабильных молекул (например, углеводородов). Представляется, что этот метод имеет некоторые перспективы для исследования CVD процессов, основанных на использовании металлорганических соединений в качестве реагентов. Оптические методы
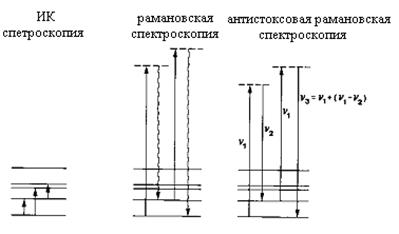
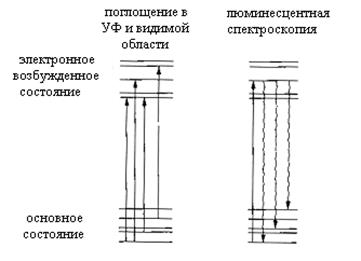
Оптические методы диагностики нашли наиболее широкое применение для диагностики состава реакционной газовой фазы, образующейся в зоне осаждения CVD реакторов. Одной из основных достоинств этих методов состоит в том, что они вносят возмущения в ход процесса в значительно меньшей степени по сравнению с методами, требующими отбора пробы. Кроме того, оптические методы обеспечивают возможность детальной идентификации компонентов реакционной газовой фазы, количественное измерение их концентраций и характеризуются высоким пространственным разрешением. Недостатками оптических методов является специфичность каждого из них в отдельности. Некоторые методы могут быть использованы только при определенных условиях и для ограниченной группы газообразных компонентов, или позволяют в большей степени получать качественные результаты нежели количественные. Кроме того, необходимо иметь в виду, что при использовании фотонов больших энергий или оптического излучения высокой интенсивности (например, от мощных лазеров) могут инициироваться фотохимические реакции, искажающие реальную картину в исследуемом CVD процессе. Методы, рассматриваемые в этом разделе, относятся к хорошо разработанным спектральным методам, и могут быть разделены на две группы. К первой группе относятся методы, основанные на колебательно-вращательных переходах в молекулах, а ко второй – электронных переходах. Инфракрасная спектроскопия поглощения, лазерная рамановская спектроскопия и когерентная антистоксовая рамановская спектроскопия относятся к первой группе, а ультрафиолетовая спектроскопия поглощения и люминесцентная спектроскопия с лазерным возбуждением – ко второй группе. На рис.31 и рис.32 схематично приведены схемы оптических переходов, поясняющие природу возникновения соответствующих спектров, на которых прямыми стрелками показаны переходы, обусловленные поглощением фотонов молекулой, а волнистыми стрелками излучательные переходы, сопровождающиеся излучением молекулой фотона.
Рис.31. Схемы оптических переходов, поясняющие принципы колебательных спектроскопий.
Рис.32. Схемы оптических переходов, поясняющие принципы спектроскопий, основанных на электронных переходах в молекуле.
Инфракрасная спектроскопия поглощения. Колебательная спектроскопия объединяет спектры поглощения в инфракрасной области и спектры комбинационного рассеяния (рамановские спектры). Оба вида спектров связаны с периодическими изменениями относительного расположения атомных ядер, т.е. с колебательным движением. Если подвергнуть какое-либо вещество воздействию непрерывной световой энергии инфракрасного диапазона (10 – 5000 см-1) и разложить прошедший через вещество световой поток по длинам волн, а затем измерить энергию светового потока в зависимости от длины волны, то получится спектр поглощения (пропускания) данного вещества (рис.33).
Рис.33. Инфракрасный спектр
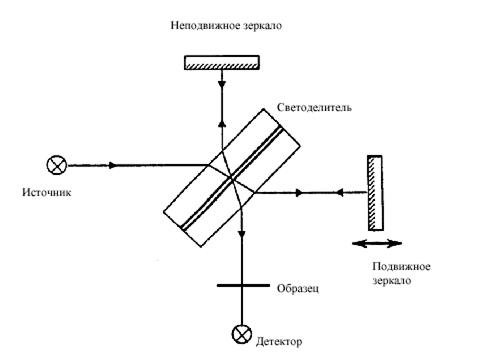
На фоне базовой линии с интенсивностью Iо появляются полосы поглощения с волновыми числами ν 1, ν 2, ν 3 и т.д. (ν - величина, обратная длине волны (в см), измеренной в вакууме. В прикладной спектроскопии волновое число обычно называют частотой. Интенсивности I этих полос различны и могут измеряться в % пропускания Т = I / Iо .100. В зависимости от состава, строения и природы химической связи спектр вещества является индивидуальным по числу полос, их положению на шкале волновых чисел и интенсивности. Полосы поглощения, наблюдающиеся как в низкочастотной (от 10 - 400 см-1), так и в других ИК областях (от 400 - 5000 см-1) имеют единую природу и интерпретируются на основе теории колебаний многоатомных систем и электрооптической теории интенсивности. ИК спектры могут быть уникальным источником информации о строении и свойствах неорганических, координационных, металлорганических соединений, полимеров и биологических объектов. По общим принципам устройства ИК спектрометров их можно разделить на две основные группы. Первая включает приборы с последовательным сканированием и регистрацией спектра с помощью одноканального приемника, а вторая – спектрометры, в которых на приемник попадает излучение всего изучаемого спектрального диапазона, но сигналы преобразуются и расшифровываются так, что можно получить информацию о каждом отдельном участке с регистрацией полного спектра во всем диапазоне. Приборы этих групп могут быть диспергирующие (призменные) и недиспергирующие. По схеме освещения ИК спектрометры бывают одно- и двухлучевые. При однолучевой схеме спектр поглощения исследуемого вещества регистрируется вместе с фоновым поглощением. Чтобы получить спектр (в процентах пропускания), нужно зарегистрировать также кривую интенсивности испускания источника (фоновый спектр). Принимая интенсивность при каждой λ в регистрируемом спектре и спектре испускания соответственно как Iλ и Iоλ , находят значение пропускания Тλ = (Iλ / Iоλ ). 100 % и строят по точкам спектральную кривую зависимости от λ (или от ν ). Обычно используется двухлучевая схема, которая позволяет выравнивать фон, т.е. линию полного пропускания, и компенсировать поглощение атмосферных паров H2O и СО2. Диспергирующие приборы первой группы – это сканирующие спектрометры. В качестве диспергирующего устройства (т.е. устройства для разложения спектра) используются призмы из монокристаллов KBr, NaCl, LiF (для средней ИК области 400 – 5000 см-1), CsI (для 400 – 200 см-1) и дифракционные решетки. Несмотря на высокое качество этих приборов, они все больше заменяются на Фурье-спектрометры, относящиеся к группе недиспергирующих приборов. В основу конструкции Фурье-спектрометров положено явление интерференции волн электромагнитного излучения. Для изготовления этих приборов используются интерферометры нескольких типов. Однако, наибольшее распространение получил интерферометр Майкельсона (рис.34). Поток инфракрасного излучения от источника преобразуется в параллельный пучок и затем разделяется на два луча приблизительно одинаковой интенсивности с помощью светоделителя. Один луч попадает на подвижное зеркало, другой – на неподвижное. Отраженные от зеркал лучи возвращаются тем же оптическим путем на светоделитель.
Рис.34. Интерферометр Майкельсона
|
Последнее изменение этой страницы: 2019-10-03; Просмотров: 285; Нарушение авторского права страницы