
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
ГАЛЬВАНИЧЕСКИЕ ЭЛЕМЕНТЫ. ЭДС.
При прохождении электрического тока через электролит на поверхности электродов протекают электрохимические реакции. Протекание электрохимических реакций может порождаться внешним источником тока. Возможно и обратное явление: электрохимические реакции, протекающие на двух электродах, опущенных в электролит, порождают электрический ток, причем реакции идут только при замкнутой цепи (при прохождении тока). Электрохимическим (или гальваническим) элементом называется устройство для получения электрического тока за счет электрохимических реакций. Простейший электрохимический элемент состоит из двух металлических электродов (проводников первого рода), опущенных в электролит (проводник второго рода) и соединенных между собой металлическим контактом. Несколько электрохимических элементов, соединенных последовательно, образуют электрохимическую цепь. Важнейшей количественной характеристикой электрохимического элемента является электродвижущая сила (ЭДС, Е), которая равна разности потенциалов правильно разомкнутого элемента (такого, у которого к конечным электродам элемента присоединены проводники первого рода из одного и того же материала). Если при прохождении электрического тока в разных направлениях на поверхности электрода протекает одна и та же реакция, но в противоположных направлениях, то такие электроды, а также элемент или цепь, составленные из них, называются обратимыми. ЭДС обратимых элементов является их термодинамическим свойством, т.е. зависит только от Т, Р, природы веществ, составляющих электроды и растворы, и концентрации этих растворов. Пример обратимого элемента - элемент Даниэля-Якоби: (-) Cu ç Zn ç ZnSO4 ç ç CuSO4 ç Cu (+) в котором каждый электрод обратим. При работе элемента идут следующие реакции: Zn ® Zn2+ + 2e, Cu2+ + 2e ® Cu. При пропускании тока бесконечно малой силы от внешнего источника на электродах протекают обратные реакции. Пример необратимого элемента - элемент Вольта: (-) Zn ç H2SO4 ç Cu (+) При работе элемента протекают реакции: Zn ® Zn2+ + 2e, 2H+ + 2e ® H2. При пропускании тока от внешнего источника электродными реакциями будут: 2H+ + 2e ® H2, Cu ® Cu2+ + 2e. ЭДС электрохимического элемента является величиной положительной, т.к. она соответствует определенному самопроизвольно протекающему процессу, дающему положительную работу. Обратному процессу, который не может протекать самостоятельно, отвечала бы отрицательная ЭДС. При составлении цепи электрохимических элементов процесс в одном из элементов можно направить так, чтобы он сопровождался затратой работы извне (несамопроизвольный процесс), используя для этого работу другого элемента цепи, в котором идет самопроизвольный процесс. Суммарная ЭДС любой цепи равна алгебраической сумме положительных и отрицательных величин. Поэтому очень важно при записи схемы цепи учитывать знаки ЭДС, пользуясь принятыми правилами. ЭДС электрохимической цепи считается положительной, если при записи цепи правый электрод заряжен положительно относительно левого (катионы при работе цепи проходят в растворе от электрода, записанного слева, по направлению к электроду, записанному справа, и в этом же направлении движутся во внешней цепи электроны). Пример.
ТЕРМОДИНАМИКА ГАЛЬВАНИЧЕСКОГО ЭЛЕМЕНТА. Пусть в электрохимической системе обратимо и изотермически протекает реакция: nAA + nBB +... ± nF Û nLL + nMM +... ± Электрическая энергия, вырабатываемая элементом, равна полезной работе А¢ суммарного процесса. Полезная работа А¢ обратимого процесса максимальна и при Р, Т = const равна убыли изобарного потенциала системы: -DGP, T = nFEP, T EP, T - обратимая ЭДС системы. EP, T = -DGP, T / nF, EV, T = -DFV, T / nF
Т.о., измерив ЭДС элемента и ее температурный коэффициент, можно найти величины DG и DS для суммарного процесса, протекающего в гальваническом элементе. Этот процесс является самопроизвольным, следовательно, DG < 0. По уравнению Гиббса-Гельмгольца можно вычислить изменение энтальпии процесса: DG = DH + T DH = DG - T nFEP = -DH + nFT nFEV = -DU + nFT Из уравнений следует, что соотношение между электрической энергией, обратимо генерируемой или поглощаемой в электрохимической системе, и тепловым эффектом протекающей в ней реакции зависит от знака и величины температурного коэффициента ЭДС dE/dT: 1. Если dE/dT > 0 , то nFE > 2. Если dE/dT < 0, то nFE < 3. Если dE/dT = 0, то DG = DH и nFE = Для расчета ЭДС уравнения можно переписать в виде: EV = EP = При использовании уравнений необходимо помнить, что они справедливы только для обратимых электрохимических систем, поэтому при изучении зависимости ЭДС от Т необходимо избегать применения электрохимических систем с жидкостными границами, т.к. возникающие на них диффузионные потенциалы не являются равновесными. Свяжем ЭДС элемента с константой равновесия реакции, протекающей в элементе. Уравнение изотермы химической реакции: -DG = RT lnKa - RT E = - Первый член правой части уравнения при заданных Р, Т - величина постоянная, его можно обозначить через Ео. Ео - стандартная ЭДС элемента (электрохимической системы) , т.е. ЭДС при всех ai = 0. Е = Ео + Т.о., ЭДС электрохимической системы является функцией активностей участников электрохимической реакции. Вышеприведенные уравнения дают возможность вычислить величины DG и Ка по экспериментальным значениям Е и, наоборот, рассчитывать Е, зная термодинамические характеристики химической реакции.
ИЗМЕРЕНИЕ ЭДС. Для измерения равновесной (обратимой) величины ЭДС электрохимического элемента необходимо, чтобы процесс совершался бесконечно медленно, т.е. чтобы элемент работал при бесконечно малой силе тока. Это условие выполняется в компенсационном методе, который основан на том, что элемент включается последовательно против внешней разности потенциалов и последняя выбирается так, чтобы ток в цепи отсутствовал. Тогда внешняя разность потенциалов равна ЭДС цепи. Пользуясь компенсационным методом, можно непосредственно измерить значение ЭДС, однако это довольно сложная операция, поэтому в лабораторной практике предпочитают сравнивать ЭДС изучаемого элемента с ЭДС так называемых стандартных (нормальных) элементов, которая тщательно измерена при разных Т. Этот сравнительный метод также является компенсационным. Основным нормальным элементом является насыщенный элемент Вестона. (Схеме измерения ЭДС - самостоятельно).
СТРОЕНИЕ ГРАНИЦЫ ЭЛЕКТРОД-РАСТВОР. ДВОЙНОЙ ЭЛЕКТРИЧЕСКИЙ СЛОЙ. При соприкосновении проводника первого рода с электролитом на границе электрод-раствор возникает двойной электрический слой. В качестве примера рассмотрим медный электрод, погруженный в раствор CuSO4. Химический потенциал ионов меди в металле при данной Т можно считать постоянным, тогда как химический потенциал ионов меди в растворе зависит от концентрации соли; в общем случае эти химические потенциалы неодинаковы. Пусть концентрация CuSO4 такова, что 2Fj = Разность электрических потенциалов и разность химических потенциалов скомпенсированы при электрохимическом равновесии. Пусть концентрация CuSO4 настолько мала, что Можно выбрать такую концентрацию электролита, при которой химические потенциалы ионов в металле и растворе одинаковы. Растворы такой концентрации получили название нулевых растворов. При погружении металла в его нулевой раствор на поверхности электрода не возникает ДЭС, однако и в этом случае разность потенциалов между металлом и раствором не равна нулю. Согласно Нернсту, единственным источником ЭДС электрохимического элемента является ДЭС на поверхности электродов. Потенциал металлов в нулевом растворе Нернст определял как абсолютный нуль потенциалов. В работах А.Н.Фрумкина было показано, что представления Нернста являются неверными. Экспериментально установлено, что ЭДС элемента, составленного их двух различных электродов, погруженных в свои нулевые растворы, весьма значительно отличается от нуля (может быть более 1 В). Потенциал металла в нулевом растворе, получивший название потенциала нулевого заряда, нельзя рассматривать как абсолютный нуль потенциалов. ТЕОРИЯ КОНДЕНСИРОВАННОГО ДВОЙНОГО СЛОЯ ГЕЛЬМГОЛЬЦА. Первую количественную теорию строения ДЭС на границе металл-раствор создал Гельмгольц (1853). По Гельмгольцу, ДЭС можно уподобить плоскому конденсатору, одна из обкладок которого совпадает с плоскостью, проходящей через поверхностные заряды в металле, другая - с плоскостью, соединяющей центры зарядов ионов в растворе, притянутых к поверхности металла электростатическими силами. Толщина двойного слоя l равна радиусу ионов r. По условию электронейтральности число притянутых к поверхности металла ионов должно быть таким, чтобы их заряды компенсировали поверхностные заряды металла, т.е. qM = - qL Теория конденсированного двойного слоя позволяет получить значения емкости ДЭС, согласующиеся с опытом, и физически правдоподобную толщину ДЭС. Однако она не может истолковать многие опытные закономерности: экспериментально найденные значения электрокинетического потенциала (x-потенциала) и их зависимость от концентрации электролита, изменение знака заряда поверхности металла в присутствии ПАВ. ТЕОРИЯ ДИФФУЗНОГО ДВОЙНОГО СЛОЯ ГУИ-ЧАПМАНА. В теории Гельмгольца не учитывается, что свойства ДЭС изменяются с концентрацией электролита и его Т. Гуи (1910) и Чапман (1913) попытались связать плотность заряда в ДЭС с составом раствора. Они учли, что помимо электростатических сил, возникающих между металлом и ионами, на ионы также действуют силы теплового молекулярного движения. При наложении этих двух сил ионы в растворе должны распределяться относительно поверхности металла диффузно - с убывающей при удалении от нее объемной плотностью заряда. Гуи и Чапман считали, что ионы можно рассматривать как материальные точки, не имеющие собственного объема, но обладающие зарядом, и что их распределение в поле заряда электрода подчиняется распределению Больцмана. Теория Гуи-Чапмана лучше теории Гельмгольца согласуется с закономерностями электрокинетических явлений. Если предположить, что начиная с некоторого расстояния l1 ионы уже не связаны прочно с поверхностью электрода при относительном перемещении твердой и жидкой фаз, то соответствующий этому расстоянию потенциал можно считать x-потенциалом (x < j). Однако теория не объясняет изменение знака x-потенциала и перезарядку поверхности с изменением состава раствора. Кроме того, теория Гуи-Чапмана оказывается менее удовлетворительной, чем теория Гельмгольца, при использовании ее для количественных расчетов емкости ДЭС, т.к. она не учитывает собственного объема ионов, которые отождествляются с материальными точками. Т.о., теория Гуи-Чапмана оправдывается лучше всего там, где теория Гельмгольца оказывается неприложимой, и, наоборот, последняя дает лучшую сходимость с опытом в тех случаях, когда первая дает неверные результаты. Следовательно, строению ДЭС должно отвечать некоторое сочетание моделей, предложенных Гельмгольцем и Гуи-Чапманом. Такое предположение было сделано Штерном (1924) в его адсорбционной теории ДЭС. АДСОРБЦИОННАЯ ТЕОРИЯ ШТЕРНА. Штерн полагал, что определенная часть ионов удерживается вблизи поверхности раздела металл-электролит, образуя гельмгольцевскую или конденсированную обкладку двойного слоя с толщиной, отвечающей среднему радиусу ионов электролита. Остальные ионы, входящие в ДЭС, распределяются диффузно с постепенно убывающей плотностью заряда. Для диффузной части ДЭС Штерн, как и Гуи, пренебрег собственными размерами ионов. Кроме того, Штерн высказал мысль, что в плотной части ДЭС ионы удерживаются за счет не только электростатических сил, но и сил специфической адсорбции, т.е. силами некулоновского происхождения. Поэтому в растворах, содержащих поверхностно-активные ионы, их число в плотной части ДЭС может превосходить заряд поверхности металла на некоторую величину, зависящую от свойств ионов и заряда металла. Т.о., по Штерну, следует различать две модели ДЭС, одна из которых относится к растворам поверхностно-инактивных электролитов, другая - к растворам, содержащим специфически адсорбирующиеся ионы. В адсорбционной теории также сохраняется равенство: - qM = qL = q1 + q2 Плотность заряда со стороны раствора qL состоит из двух частей: плотности заряда в гельмгольцевском слое q1 и плотности заряда в диффузном слое q2. Теория Штерна позволяет определить x-потенциал как падение потенциала в диффузной части ДЭС, где уже потеряна прочная связь между металлом и ионами. При таком определении x-потенциал не должен совпадать с нерстовским потенциалом, как это и наблюдается на опыте. Теория Штерна смогла объяснить и перезарядку поверхности твердого тела. При бесконечно малой концентрации все заряды в растворе распределены диффузно, и строение ДЭС описывается теорией Гуи-Чапмана. Напротив, в концентрированных растворах строение ДЭС приближается к модели, предложенной Гельмгольцем. В области средних концентраций, где x сравним по величине с RT/F, его зависимость от концентрации можно выразить приближенными уравнениями: для положительных величин x: x = В - для отрицательных значений x: x = В¢ + Теория Штерна дает качественно правильную картину ДЭС. Определение емкости с использованием модели Штерна согласуется с опытом как по величинам емкости, так и по характеру ее зависимости от потенциала электрода и концентрации раствора. Но теория Штерна не свободна от недостатков. К их числу относится невозможность количественного описания емкостных кривых, особенно при удалении от потенциала нулевого заряда. ДАЛЬНЕЙШЕЕ РАЗВИТИЕ ТЕОРИИ СТОЕНИЯ ДЭС. Было предпринято много попыток разработать теорию ДЭС, количественно согласующуюся с опытными данными (Райс, Фрумкин с сотр., Бокрис, Деванатхан, Есин, Мюллер, Парсонс, Эршлер и др.). Наибольшее признание получила модель Грэма (1947). Согласно Грэму, обкладка ДЭС, находящаяся в растворе, состоит не из двух, а из трех частей. Первая, считая от поверхности металла, называется внутренней плоскостью Гельмгольца; в ней находятся лишь поверхностно-активные ионы (заряд плоскости равен q1) либо, если их нет в растворе, молекулы растворителя (q1 = 0); потенциал ее, отнесенный к раствору, обозначается y1. Следующая, удаленная от поверхности металла на расстояние, до которого могут подходить ионы (центры их заряда), называется внешней плоскостью Гельмгольца; ее общий заряд равен q2, а потенциал плоскости y2. За внешней плоскостью Гельмгольца располагается диффузный слой с потенциалом, изменяющимся от y2 до нуля и с плотностью заряда, совпадающей с q2. Модель Грэма отражает основные черты и особенности структуры ДЭС металл-электролит. Она позволяет рассчитать кривые дифференциальной емкости для любых концентраций данного электролита, если имеется экспериментальная кривая хотя бы для одного его раствора. Однако и эта модель охватывает далеко не все аспекты проблемы.
Лекция 46 Популярное:
|
Последнее изменение этой страницы: 2016-03-15; Просмотров: 2197; Нарушение авторского права страницы

 = -DS = -nF
= -DS = -nF 


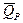 + nFT
+ nFT  =
=  + nFT
+ nFT  + T
+ T  + T
+ T 
 =
=  lnKa -
lnKa -  = Eo + 2, 303
= Eo + 2, 303  >
>  . Тогда при погружении металла в раствор часть ионов Cu2+ из раствора дегидратируется и перейдет на металл, создав на нем положительный заряд. Этот заряд будет препятствовать дальнейшему переходу ионов Cu2+ из раствора на металл и приведет к образованию вблизи электрода слоя притянутых к нему анионов SO42-. Установится так называемое электрохимическое равновесие, при котором химические потенциалы ионов в металле и в растворе будут отличаться на величину разности потенциалов образующегося при этом двойного электрического слоя (ДЭС):
. Тогда при погружении металла в раствор часть ионов Cu2+ из раствора дегидратируется и перейдет на металл, создав на нем положительный заряд. Этот заряд будет препятствовать дальнейшему переходу ионов Cu2+ из раствора на металл и приведет к образованию вблизи электрода слоя притянутых к нему анионов SO42-. Установится так называемое электрохимическое равновесие, при котором химические потенциалы ионов в металле и в растворе будут отличаться на величину разности потенциалов образующегося при этом двойного электрического слоя (ДЭС):  lnс
lnс