
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Недостатки теории Аррениуса. Теория электролитов Дебая и Гюккеля.
Активность растворенной соли а может быть определена по давлению пара, температуре затвердевания, по данным о растворимости, методом ЭДС. Все методы определения активности соли приводят к величине, характеризующей реальные термодинамические свойства растворенной соли в целом, независимо от того, диссоциирована она или нет. Однако в общем случае свойства различных ионов неодинаковы, и можно ввести и рассматривать термодинамические функции отдельно для ионов разных видов: m+ = m+о + RT ln a+ = m+o + RT ln m+ + RT ln g+¢ m- = m-о + RT ln a- = m-o + RT ln m- + RT ln g-¢ где g+¢ и g-¢ - практические коэффициенты активности (коэффициенты активности при концентрациях, равных моляльности m). Но термодинамические свойства различных ионов не могут быть определены порознь из опытных данных без дополнительных допущений; мы можем измерить только средние термодинамические величины для ионов, на которые распадается молекула этого вещества. Пусть диссоциация соли происходит по уравнению: Аn+Вn- = n+ Аz+ + n- Bz- При полной диссоциации m+ = n+ m, m- = n- m. Пользуясь уравнениями Гиббса-Дюгема, можно показать: а+n+× а-n-¤ а = const Стандартные состояния для нахождения величин активностей определяются так: lim a+ ® m+ = n+ m при m ® 0, lim a- ® m- = n- m при m ® 0 Стандартное состояние для а выбирается так, чтобы const была равна 1. Тогда: а+n+× а-n- = а Т.к. нет методов экспериментального определения значений а+ и а- в отдельности, то вводят среднюю ионную активность а±, определяемую соотношением: а±n = а Т.о., мы имеем две величины, характеризующие активность растворенной соли. Первая из них - это мольная активность, т.е. активность соли, определяемая независимо от диссоциации; она находится теми же экспериментальными методами и по тем же формулам, что и активность компонентов в неэлектролитах. Вторая величина - средняя ионная активность а±. Введем теперь коэффициенты активности ионов g+¢ и g-¢ , среднюю ионную моляльность m± и средний ионный коэффициент активности g±¢ : a+ = g+¢ m+, a- = g-¢ m-, m± = (m+n+× m-n-)1/n = (n+n+× n-n-)1/n m g±¢ = (g¢ +n+× g¢ -n-)1/n Очевидно: a±= (g¢ +n+× g¢ -n-)1/n (n+n+× n-n-)1/n m = g±¢ m± Т.о., основные величины связаны соотношениями: a± = g±¢ m± = g±¢ (n+n+× n-n-)1/n m = L g±¢ m где L = (n+n+× n-n-)1/n и для солей каждого определенного типа валентности является величиной постоянной. Величина g±¢ является важной характеристикой отклонения раствора соли от идеального состояния. В растворах-электролитах, как и в растворах-неэлектролитах, могут быть использованы следующие активности и коэффициенты активности: g± = g±¢ = f± = Основными методами измерения величины g±¢ являются криоскопический и метод ЭДС. Многочисленные исследования показали, что кривая зависимости g±¢ от концентрации раствора (m) имеет минимум. Если изображать зависимость в координатах lg g±¢ - Присутствие в растворе других солей изменяет коэффициент активности данной соли. Суммарное влияние смеси солей в растворе на коэффициент активности каждой из них охватывается общей закономерностью, если суммарную концентрацию всех солей в растворе выразить через ионную силу. Ионной силой I (или ионной крепостью) раствора называется полусумма произведений концентрации каждого иона на квадрат числа его заряда (валентности), взятая для всех ионов данного раствора. Если использовать моляльность как меру концентрации, то ионная сила раствора определяется выражением: I = где i - индексы ионов всех солей в растворе; mi = ni m. Льюис и Рендалл открыли эмпирический закон ионной силы: средний ионный коэффициент активности g±¢ диссоциирующего на ионы вещества является универсальной функцией ионной силы раствора, т.е. в растворе с данной ионной силой все диссоциирующие на ионы вещества имеют коэффициенты активности, не зависящие от природы и концентрации данного вещества, но зависящие от числа и валентности его ионов. Закон ионной силы отражает суммарное взаимодействие ионов раствора с учетом их валентности. Этот закон точен лишь при очень малых концентрациях (m ≤ 0, 02); уже при умеренных концентрациях он верен лишь приблизительно. В разбавленных растворах сильных электролитов: lg g±¢ = - А НЕДОСТАТКИ ТЕОРИИ АРРЕНИУСА. В теории электролитов очень важным является вопрос о распределении ионов в растворе. По первоначальной теории электролитической диссоциации, основанной на физической теории растворов Вант-Гоффа, считалось, что ионы в растворах находятся в состоянии беспорядочного движения - в состоянии, подобном газообразному. Однако представление о беспорядочном распределении ионов в растворе не соответствует действительности, так как онo не учитывают электростатического взаимодействия между ионами. Электрические силы проявляются на относительно больших расстояниях, и в сильных электролитах, где диссоциация велика, а концентрация ионов значительна и расстояния между ними невелики, электростатическое взаимодействие между ионами настолько сильно, что оно не может не сказываться на характере их распределения. Возникает тенденция к упорядоченному распределению, аналогичному распределению ионов в ионных кристаллах, где каждый ион окружён ионами противоположного знака. Распределение ионов будет определяться соотношением электростатической энергии и энергии хаотического движения ионов. Эти энергии сравнимы по величине, поэтому реальное распределение ионов в электролите является промежуточным между беспорядочным и упорядоченным. В этом заключается своеобразие электролитов и трудности, возникающие при создании теории электролитов. Около каждого иона образуется своеобразная ионная атмосфера, в которой преобладают ионы противоположного (по сравнению с центральным ионом) знака. Теория Аррениуса не учитывала этого обстоятельства, и многие выводы этой теории оказались в противоречии с опытом. В качестве одной из количественных характеристик электролита теория Аррениуса предлагает степень электролитической диссоциации a, определяющую долю ионизированных молекул в данном растворе. В согласии с ее физическим смыслом a не может быть больше 1 или меньше 0; при заданных условиях она должна быть одной и той же, независимо от метода ее измерения (по измерению электропроводности, осмотического давления или ЭДС). Однако на практике значения a, полученные разными методами, совпадают только для разбавленных растворов слабых электролитов; для сильных электролитов расхождение тем больше, чем больше концентрация электролита, причем в области высоких концентраций a становится больше 1. Следовательно, a не может иметь того физического смысла, который ей приписывался теорией Аррениуса. Второй количественной характеристикой по теории Аррениуса является константа диссоциации; она должна быть постоянной для данного электролита при заданных Т и Р, независимо от концентрации раствора. На практике только для разбавленных растворов очень слабых электролитов Кдис остается при разбавлении более или менее постоянной. Т.о., теория электролитической диссоциации приложима только к разбавленным растворам слабых электролитов. ТЕОРИЯ ЭЛЕКТРОЛИТОВ ДЕБАЯ И ГЮККЕЛЯ. Основные положения современной теории растворов электролитов были сформулированы в 1923 г. Дебаем и Гюккелем. Для статистической теории электролитов исходным является следующее положение: ионы распределены в объеме раствора не хаотически, а в соответствии с законом кулоновского взаимодействия. Вокруг каждого отдельного иона существует ионная атмосфера (ионное облако) - сфера, состоящая из ионов противоположного знака. Ионы, входящие в состав сферы, непрерывно обмениваются местами с другими ионами. Все ионы раствора равноценны, каждый из них окружен ионной атмосферой, и в то же время каждый центральный ион входит в состав ионной атмосферы какого-либо другого иона. Существование ионных атмосфер и есть тот характерный признак, который, по Дебаю и Гюккелю, отличает реальные растворы электролитов от идеальных. С помощью уравнений электростатики можно вывести формулу дляэлектрического потенциала ионной атмосферы, из которой вытекают уравнения для средних коэффициентов активности в электролитах: Y = D - диэлектрическая проницаемость раствора; е - заряд электрона; zi - заряд иона; r - координата (радиус). c = Величина 1/c называется характеристической длиной ; ее можно отождествить с радиусом ионной атмосферы. Она имеет большое значение в теории растворов электролитов. Для коэффициента активности получено следующее выражение: lg f± = - A |z+× z-| Коэффициент A зависит от Т и D: обратно пропорционален (DT)3/2. Для водных растворов 1-1 зарядных электролитов при 298 К, допуская равенство диэлектрических проницаемостей раствора и растворителя (78, 54), можно записать: lg f± = - A Т.о., теория Дебая и Гюккеля позволяет получить такое же уравнение для коэффициента активности, какое было эмпирически найдено для разбавленных растворов электролитов. Теория, следовательно, находится в качественном согласии с опытом. При разработке этой теории были сделаны следующие допущения: 1. Число ионов в электролите можно определить из аналитической концентрации электролита, т.к. он считается полностью диссоциированным (a = 1). Теорию Дебая и Гюккеля поэтому иногда называют теорией полной диссоциации. Однако ее можно применить и в тех случаях, когда a ¹ 1. 2. Распределение ионов вокруг любого центрального иона подчиняется классической статистике Максвелла-Больцмана. 3. Собственными размерами ионов можно пренебречь по сравнению с расстояниями между ними и с общим объемом раствора. Т.о., ионы отождествляются с материальными точками, и все их свойства сводятся лишь к величине заряда. Это допущение справедливо только для разбавленных растворов. 4. Взаимодействие между ионами исчерпывается кулоновскими силами. Наложение сил теплового движения приводит к такому распределению ионов в растворе, для которого характерна статистическая шаровая ионная атмосфера. Это допущение справедливо лишь для разбавленных растворов. При повышении концентрации среднее расстояние между ионами уменьшается, и наряду с электростатическими силами появляются другие силы, действующие на более близком расстоянии, в первую очередь силы Ван-дер-Ваальса. Возникает необходимость учета взаимодействия не только между данным ионом и его окружением, но и между любыми двумя соседними ионами. 5. При расчетах принимается, что диэлектрические проницаемости раствора и чистого растворителя равны; это справедливо только в случае разбавленных растворов. Т.о., все допущения Дебая и Гюккеля приводят к тому, что их теория может быть применима только к разбавленным растворам электролитов с ионами низкой валентности. Уравнение (1) соответствует этому предельному случаю и выражает так называемый предельный закон Дебая и Гюккеля или первое приближение теории Дебая и Гюккеля. Предельный закон Дебая-Гюккеля дает верные значения коэффициентов активности 1-1 зарядного электролита, особенно в очень разбавленных растворах. Сходимость теории с опытом ухудшается по мере увеличения концентрации электролита, увеличения зарядов ионов и уменьшения диэлектрической проницаемости растворителя, т.е. с ростом сил взаимодействия между ионами. Первая попытка усовершенствовать теорию Дебая и Гюккеля и расширить область ее применения была сделана самими авторами. Во втором приближении они отказались от представления об ионах как о материальных точках (допущение 3) и попытались учесть конечные размеры ионов, наделив каждый электролит некоторым средним диаметром а (при этом изменяется и допущение 4). Приписав ионам определенные размеры, Дебай и Гюккель учли тем самым силы некулоновского происхождения, препятствующие сближению ионов на расстояние, меньшее некоторой величины. Во втором приближении средний коэффициент активности описывается уравнением: lg f± = - где А сохраняет прежнее значение; а условно названо средним эффективным диаметром ионов , имеет размерность длины, фактически - эмпирическая постоянная; В = c/ Сохранив основные положения второго приближения теории, Гюккель учел уменьшение диэлектрической проницаемости с ростом концентрации растворов. Ее уменьшение вызывается ориентацией диполей растворителя вокруг иона, в результате чего снижается их реакция на эффект внешнего поля. Уравнение Гюккеля выглядит следующим образом: lg f± = - где С - эмпирическая константа. При удачном подборе значений В и С формула Гюккеля хорошо согласуется с опытом и широко используется при расчетах. При последовательном уменьшении ионной силы уравнение (3) последовательно переходит в формулу второго приближения теории Дебая и Гюккеля (уравнение (2)), а затем в предельный закон Дебая-Гюккеля (уравнение (1)). В процессе развития теории Дебая-Гюккеля и последовательного отказа от принятых допущений улучшается сходимость с опытом и расширяется область ее применимости, однако это достигается ценой превращения теоретических уравнений в полуэмпирические.
Лекция 44 Популярное:
|
Последнее изменение этой страницы: 2016-03-15; Просмотров: 2264; Нарушение авторского права страницы
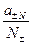 - рациональный коэффициент активности (практически не применяется);
- рациональный коэффициент активности (практически не применяется);  - практический коэффициент активности (средний моляльный);
- практический коэффициент активности (средний моляльный);  - средний мольный коэффициент активности.
- средний мольный коэффициент активности.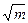 , то для разбавленных растворов зависимость оказывается линейной. Наклон прямых, соответствующих предельному разбавлению, одинаков для солей одного валентного типа.
, то для разбавленных растворов зависимость оказывается линейной. Наклон прямых, соответствующих предельному разбавлению, одинаков для солей одного валентного типа.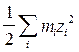

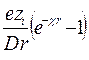
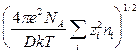 - величина, зависящая от концентрации раствора, D и Т, но не зависящая от потенциала; имеет размерность обратной длины; характеризует изменение плотности ионной атмосферы вокруг центрального иона с увеличением расстояния r от этого иона.
- величина, зависящая от концентрации раствора, D и Т, но не зависящая от потенциала; имеет размерность обратной длины; характеризует изменение плотности ионной атмосферы вокруг центрального иона с увеличением расстояния r от этого иона.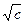 = - 0, 51
= - 0, 51 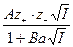 (2)
(2)