
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Промышленные термические процессы
Теоретические разработки механизма термических процессов, происходящие при переработке нефти легли в основу технологического оформления ряда промышленных процессов. 8.6.1. Термический крекинг. К чисто термическому процессу относится термический крекинг, первая промышленная установка которого была пущена в 1912 г. с целью получения автобензина. Однако из-за постоянного ужесточения требований к качеству бензинов за последние 50 лет термический крекинг для этой цели уже не применяется, и классическая технология этого процесса перестала существовать. Эксплуатируемые в настоящее время установки «легкого» крекинга служат для получения вакуумного газойля и маловязкого котельного топлива. Современные крекинг-процессы включают, как правило, комплексную переработку тяжелого нефтяного сырья и содержат установки легкого крекинга (висбрекинга), при 400 – 450 оС, каталитического крекинга, гидроочистки, производства оксигенатов и т. д. 8.6.2. Промышленный пиролиз . Как уже упоминалось ранее, он предназначен для получения низших алкенов (этилена и пропилена), а также 1, 3-бутадиена, стирола и некоторых других продуктов. Сырьем для процесса служат разнообразные нефтепродукты. В США – это главным образом газообразные углеводороды. В России, странах СНГ, Западной Европе, наряду с газообразными алканами, применяют бензиновые и керосино-газойлевые фракции. При пиролизе бензиновых фракций примерный состав продуктов процесса следующий, % масс.: метан 12 -16; этилен 22 – 32; пропилен 10 – 17; фракция С4 5 – 12; арены С6 – С8 6 – 13; смолы 4 – 8. Кроме того, в процессе образуются водород, фракция С5 и др. При пиролизе керосино-газойлевых фракций выход метана и этилена существенно снижается, пропилена почти не изменяется, но зато значительно возрастает доля жидких продуктов, в т. ч. смол, причем общее количество жидких продуктов может достигать 50 %. Для повышения выхода низших олефинов из керосино-газойлевых фракций до уровня выхода из газообразных алкенов керосино-газойлевые фракции необходимо подвергнуть предварительной гидрообработке (гидроочистке, гидрокрекингу, гидродеароматизации). Эти виды каталитической обработки снижают количество ароматики в сырье, что приводит также к пониженному коксованию сырья. 8.6.3. Коксование . Промышленные процессы коксования проводят с использованием остаточного нефтяного сырья: мазута, полугудронов, гудронов, тяжелой пиролизной смолы, крекинг-остатков и др. Промышленностью реализованы три различные технологии: Наибольшее распространение получил второй способ, который ведут при 480–520 оС и давлении 0, 2–0, 3 МПа. Продукты процесса – кокс, газ, бензин, легкий и тяжелый газойли. Соотношение продуктов процесса зависит от состава сырья, параметров процесса и аппаратурного оформления.
Контрольные вопросы 1. Дайте сравнительную характеристику устойчивости углеводородов различных классов с повышением температуры. 2. Объясните основные положения теории радикально-цепного механизма термических реакций углеводородов, согласно теории Н. Н. Семенова. 3. В чем суть стадии инициирования цепи? Дайте определение понятия 4. По каким механизмам могут образовываться радикалы? Приведите ряд стабильности различных радикалов по мере ее снижения. 5. Приведите реакции, в которые вступают радикалы. Укажите их тепловые эффекты. 6. Приведите реакции углеводородов, приводящие к обрыву цепи. 7. Приведите условия и механизм термических превращений алканов. 8. Приведите условия и механизм термических превращений циклоалканов. 9. Приведите условия и механизм термических превращений алкенов. 10. Приведите условия и механизм термических превращений алкадиенов и алкинов. 11. Приведите условия и механизм термических превращений аренов. 12. Объясните особенности процесса пиролиза углеводородов и его задачи. 13. Объясните особенности процесса коксования углеводородов и его задачи.
ТЕМА 9. Термокаталитические реакции углеводородов
В настоящее время доля каталитических процессов в нефтепереработке и нефтехимии составляет свыше 80 % от всей продукции. При этом следует отметить, что в новейших процессах их вклад еще больше – 90 %. Основные каталитические процессы в нефтепереработке – это каталитический крекинг, каталитический риформинг, гидроочистка, гидрокрекинг, алкилирование, изомеризация и др.
Основные понятия о катализе и катализаторах По механизму взаимодействия реагирующих веществ с катализатором и по типу промежуточных продуктов различают окислительно-восстановительные (гидрирующе-дегидрирующие) и кислотно-основные катализаторы. Многие каталитические системы являются бифункциональными. Примером может служить катализатор риформинга, в котором платина несет окислительно-восстановительную функцию, а носитель платины – оксид алюминия – кислотно-основную. Кроме того, многие оксиды и сульфиды металлов обладают обеими этими функциями. Наибольшее место в нефтепереработке занимает гетерогенный катализ, осуществляемый с помощью активной поверхности твердых соединений. 9.1.1. Основные свойства катализаторов . Из химии известно, что одна и та же реакция может осуществляться на разных катализаторах и даже без них. Отличие заключается лишь в том, с какой скоростью протекает реакция в каждом таком случае. Например, если принять скорость гидрирования этилена по реакции Н2С = СН2 + Н2 = С2Н6 на хромовом катализаторе за единицу, то скорость на никелевом катализаторе составит 13, платиновом – 100, палладиевом – 1000, а на родиевом – 1800. Отсюда следует, что в данном случае родий (Rh) обладает наибольшей активностью. Обычно каталитические процессы протекают по двум и более направлениям. В связи с этим перед катализатором ставится требование обеспечить протекание процесса с наибольшим выходом целевого продукта. Способность катализатора выполнять такую задачу и называется селективностью. Стабильностью катализатораназывают способность его сохранять активность в течение длительного времени. В гомогенных процессах катализаторы, как правило, менее стабильны, чем в гетерогенных, т. к. в первых наблюдается разбавление катализатора продуктами процесса. Причины снижения активности гетерогенных катализаторов более разнообразны. Они связаны как с физическими, так и с химическими факторами. При длительном воздействии на катализатор повышенных температур и давлений может происходить рекристаллизация катализаторов, приводящая к снижению его удельной поверхности и числа активных центров. Для повышения стойкости к рекристаллизации в катализатор вводят структурообразующие добавки – промоторы. Например, в серебряный катализатор реакции: Ag
O в качестве промотирующей добавки вводят в микроколичествах фосфор, мышьяк или селен. Кроме того, физические изменения катализатора связаны с механическими воздействиями (например, алюмосиликатный катализатор каталитического крекинга истирается при движении по катализаторопроводам, а также за счет трения частиц друг о друга в псевдоожиженном слое). Химические изменения катализатора связаны с хемосорбцией на его поверхности примесей, содержащихся в исходном сырье и/или некоторыми продуктами реакции. Такие вещества называют каталитическими ядами. Это могут быть соединения серы, азота, других гетероатомов, а также металлоорганика. Кроме того, со временем происходит закоксовывание катаЛизатора, в результате чего его поверхность экранируется коксом, и активность катализатора снижается. Активность закоксованного катализатора может быть восстановлена путем обработки его кислородом воздуха или водяным паром. 9.1.2. Механизм действия окислительно-восстановительного катализатора. Специфика каталитических процессов заключается в том, что обмен электронами между реагирующими веществами протекает с участием электронов катализатора. Типичными катализаторами этого вида являются переходные металлы (Fe, Co, Ni, Ru, Rh, Pt, Re, Ir и др.) и полупроводники ( Ge, P, Si, оксиды Ni, W, Mo и др.). Активность переходных металлов заключается в незавершенности их d-орбиталей. Неспаренные электроны этой орбитали выступают как «свободная валентность» подобно свободному радикалу. Если адсорбирующаяся на поверхности металла молекула имеет свободные орбитали, то возникает донорно-акцепторная связь за счет перехода электрона из металла на вакантные уровни молекулы. Если поверхность металла обладает большим сродством к электрону, то электрон молекулы переходит к металлу. Между этими крайними случаями возможны промежуточные механизмы. Предположительно образующиеся поверхностные хемосорбированные вещества подобны комплексным веществам. В случае полупроводниковых катализаторов свободные валентности (неспаренные электроны и электронные дырки) возникают из-за неполной координации атомов поверхности кристаллической решетки и различных дефектов самого кристалла (например, узел кристалла, где отсутствует катион, ведет себя как отрицательный заряд, отталкивая электроны в ближайших к нему узлах, в силу чего эти электроны могут быть вытеснены из валентной зоны в зону проводимости). Появление электронов или дырок в зоне проводимости возможно также по причине наличия в кристалле примесей, обладающих электроннодонорными и электроноакцепторными свойствами, а также нарушением стехиометрического состава. На поверхности кристалла электроны или дырки играют роль свободной валентности или активных центров. Молекулы углеводорода, реагируя со свободными электронами (дырками) поверхности катализатора, подвергаются диссоциации. В результате диссоциации одна часть молекулы связывается с катализатором прочной двухэлектронной связью, а другая – слабой одноэлектронной. Эти связи могут взаимно переходить друг в друга. Нестабильность и высокая реакционная способность таких поверхностных соединений определяют высокую скорость их дальнейших превращений. Каждая молекула может реагировать с данным активным центром по-разному. В одном элементарном каталитическом акте могут принимать участие группы из двух, трех и более центров (дублеты, триплеты, мультиплеты). Таким образом, активность катализатора зависит от числа свободных валентностей на поверхности металла. 9.1.3. Кислотно-основной катализ . Жидкие и твердые кислоты широко используют в нефтепереработке в качестве катализаторов. Их каталитические свойства связаны с образованием катионов, называемых карбкатионами или карбоний-ионами, которые являются промежуточными продуктами взаимодействия углеводородов с кислотами. Карбкатионы образуются при передаче протона от катализатора (кислота НХ) к молекуле ненасыщенного углеводорода:
При высокой активности кислотного катализатора карбкатионы образуются также из алканов или циклоалканов путем гетеролитического разрыва связей в молекуле под воздействием катализатора (кислота Льюиса ):
Карбкатионы обладают очень высокой реакционной способностью. Константы скорости ионных реакций на несколько порядков выше аналогичных радикальных. В табл. 9.1 приведены значения стандартных теплот образования некоторых, наиболее характерных карбкатионов. Анализ данных табл. 9.1 показывает, что наибольшей реакционной способностью обладает фенильный карбкатион +С6Н5, следовательно, он же имеет наименьшую стабильность. Другой вывод состоит в том, что третичные карбкатионы более стабильны, чем вторичные, а те, в свою очередь, стабильнее первичных карбкатионов. Таблица 9.1 Стандартные теплоты образования карбкатионов
Основные реакции карбкатионов, как и радикалов – мономолекулярный распад по Наиболее рапространенные кислотные катализаторы – это алюмосиликаты, галогениды Al, B, Sb; Al2O3, сульфиды некоторых переходных металлов, а также ряд протонных кислот, например Н2SO4, H3PO4 и др. Эти кислоты применяют в процессах риформинга, каталитического крекинга, алкилирования, изомеризации и т. д..
Реакции карбкатионов 9.2.1. Изомеризация может протекать при переносе как гидрид-иона, так и металл-аниона:
9.2.2. Распад по + +
Вероятность распада по + +
Н3С СН3 СН3 СН3
Из сравнения термодинамики распада и изомеризации карбкатионов следует, что в большинстве случаев распаду должна предшествовать изомеризация. 9.2.3.Присоединение карбкатионов к алкенам и аренам. Эти реакции являются обратными распаду карбкатионов: + +
Реакции (9.8) и (9.9) идут с тепловыми эффектами, равными по абсолютной величине с реакциями распада, но противоположными по знаку. 9.2.4. Передача протона молекуле алкена или аниону катализатора осуществляется, например, в реакции: + +
СН3 СН3
Термодинамически наиболее вероятны те реакции, в которых протон отщепляется от первичного карбкатиона, а в результате образуется третичный карбкатион. При передаче протона катализатору цепь обрывается.
9.2.5. Отрыв гидрид-иона от молекулы углеводорода оуществляется, например, в реакции: +
-Н+ (9.11)
Таким путем осуществляется передача цепи. Активность карбкатиона при реакции отрыва гидрид-иона от молекулы изменяется так:
R + перв. > R + втор. > R + трет.
Карбкатионные реакции протекают либо в жидкой фазе, либо на поверхности твердого катализатора. Сольватация в растворе и адсорбция на поверхности твердого катализатора изменяют тепловые эффекты реакций. При этом теоретические соотношения тепловых эффектов реакций разных карбкатионов реально могут значительно отличаться от расчетных соотношений.
Каталитический крекинг Это процесс каталитического деструктивного превращения тяжелых фракций нефти в моторное топливо и сырье для нефтехимии, технического углерода и кокса. Процесс ведут на алюмосиликатном катализаторе при 480 – 530 оСи давлении 0, 3 – 0, 7 МПа. 9.3.1. Химические основы процесса. Основными реакциями каталитического крекинга являются: 1) расщепление высокомолекулярных углеводородов; 2) изомеризация; 3) дегидрирование циклоалканов в арены. Реализация этих реакций приводит к получению карбюраторных топлив с высоким октановым числом, т. к. октановые числа изоалканов выше, чем у н-алканов, а у аренов – чем у циклоалканов и алканов. 9.3.2. Превращения алканов . Они подвергаются изомеризации и распаду на алканы и алкены меньшей молекулярной массы, чем исходные алканы. Первая стадия процесса – зарождение цепи – происходит по двум механизмам. По первому – часть молекул алканов термически крекируется. Образовавшиеся алкены отрывают протоны от катализатора и превращаются в карбкатионы: +
По второму – образование карбкатиона происходит непосредственно из алкана путем отщепления от молекулы гидрид-иона под действием протонного центра или апротонного катализатора:
или
Так как отрыв гидрид-иона от третичного атома легче всего, изоалканы крекируются быстрее, чем н-алканы. Термодинамика процесса такая, что, превращение например, карбкатиона С7Н15+ состоит в цепи ряда последовательно-параллельных реакций изомеризации и
СН3(СН2)5СН2+
изомеризация
Так как распад алкильных карбкатионов с образованием первичных и вторичных ионов С1 – С3 происходит труднее, чем с образованием третичных ионов с большим числом атомов углерода, то скорость крекинга растет с удлинением цепи. Например, в одинаковых условиях степень превращения составит, %: н-С5Н12 – 1; н-С7Н16 –3; н-С12Н26 -18; н-С16Н34 – 42. Легкость распада ионов с отщеплением третичных карбкатионов водет к накоплению в продуктах распада алканов с семью и более атомами углерода.
В заключение следует отметить, что скорость реакций каталитического крекинга алканов на 1–2 порядка выше, чем при термическом крекинге.
9.3.3.Превращения циклоалканов. Скорость каталитического крекинга цикланов близка к скорости крекинга алканов с одинаковым числом атомов углерода. Для циклоалканов характерны следующие реакции: 1) дециклизация с образованием алкенов и диенов; 2) дегидрирование, приводящее к получению аренов; 3) изомеризация циклов и заместителей. Глубина крекинга цикланов растет с увеличением числа заместителей в кольце (табл. 9.2). Таблица 9.2 Зависимость глубины крекинга от заместителей в циклоалканах
Возрастание глубины крекинга, как показано в табл. 9.2, связано с тем, что отщепление гидрид-иона с образованием одновременно карбкатиона протекает легче с третичным атомом углерода. В то же время гемструктуры (1, 1-диметилциклогексан) отщепляют гидрид-ион от вторичного атома углерода, поэтому степень превращения в этом случае сопоставима со степенью превращения незамещенного циклогексана. Распад циклогексил-иона идет двумя способами: 1) с разрывом связи При разрыве связи С – С образуются алкены и алкадиены следующим образом: R
Алкенильный ион далее изомеризуется в аллильный: R R
Далее процесс развивается так:
или происходит передача протона алкену или катализатору: +
Распад циклогексил-карбкатиона с расщеплением связи С – Н энергетически более выгоден, так как через промежуточные циклоалкены и циклоалкины образуются арены:
- H
Циклоалкены подвергаются крекингу быстрее, чем циклоалканы:
H Н
Выход аренов составляет 25 % и более от суммы продуктов превращения циклогексанов, а газовая фаза содержит повышенное количество водорода по сравнению с газами крекинга алканов. Имеет место также изомеризация циклогексанов в циклопентаны и обратно:
Эта реакция протекает через протонированное пропановое кольцо. Равновесие в этой реакции сильно сдвинуто вправо, так как в условиях каткрекинга циклопентаны более устойчивы, чем циклогексаны. Но циклогексаны дегидрируются в арены и тем самым равновесие сдвигается влево. Вероятность превращения циклогексана в тот или иной продукт в итоге зависит от катализатора. Если в молекуле циклоалкана длинные боковые цепи, то возможна изомеризация боковой цепи и /или деалкилирование. Бицикланы ароматизируются в большей степени, чем моноциклы. Декалин превращается в нафталин на 33 % при 500 оС, тетралин – на 88 %. 9.3.4. Превращения алкено в. Скорость их каткрекинга на 2 – 3 порядка выше соответствующих алканов и циклоалканов, что объясняется термодинамикой образования из них карбкатионов: +
Вместе со многими общими реакциями с алканами алкены обладают и специфическими реакциями перераспределения водорода и циклизации. Сущность таких реакций состоит в том, что в присутствии кислых катализаторов часть алкенов теряет водород и образуются полиненасыщенные углеводороды, а другая часть алкенов гидрируется выделившимся водородом и переходит в алканы, например:
При этом сильноненасыщенные углеводороды полимеризуются, дегидрируются и, теряя водород, превращаются в кокс. Циклизация алкенов может привести к образованию циклопентанов, циклопентенов и аренов по схеме: СН3(СН2)3СН = СН2
- Н2 - 2Н2 -3Н2
метилциклопентан метициклопентен метилциклопентин бензол
Пятичленные циклы изомеризуются в шестичленные и далее ароматизуются. 9.3.5.Превращения аренов. Незамещенные арены при каталитическом крекинге стабильны. Монометилзамещенные арены по реакционной способности сравнимы с алканами, арены с более длинными алкильными заместителями по стабильности близки к алкенам. Основной реакцией для них является деалкилирование. Это объясняется тем, что ароматическое кольцо обладает большим сродством к протону по сравнению с алкил-ионом: H
в свою очередь:
Скорость реакции возрастает с увеличением длины алкильного заместителя, а также в ряду: С6Н5 – Rперв. < С6Н5 – Rвтор. < С6Н5 – Rтрет. Именно такой ряд обусловлен различной устойчивостью образующихся карбкатионов. Для метилзамещенных аренов отщепление карбкатиона затруднено, и протекают в основном реакции диспропорционирования: |
Последнее изменение этой страницы: 2017-04-13; Просмотров: 492; Нарушение авторского права страницы
 -связи на примерах алкена, алкина, циклоалкана, арена. В чем ее особенность, по сравнению с другими связями С – С и С – Н?
-связи на примерах алкена, алкина, циклоалкана, арена. В чем ее особенность, по сравнению с другими связями С – С и С – Н? 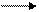 СН2 = СН2 + О2 СН2 – СН2. (9.1)
СН2 = СН2 + О2 СН2 – СН2. (9.1)

 НХ + СН3СН = СНR CH3CH2CH+R + X-. (9.2)
НХ + СН3СН = СНR CH3CH2CH+R + X-. (9.2)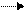 CnH2n + 2 + L (CnH2n + 1)+ + LH-. (9.3)
CnH2n + 2 + L (CnH2n + 1)+ + LH-. (9.3)




 Н
Н
 .. +
.. +

 +СН2СНСН2СН3 СН3СНСН2СН3 + 79 кДж/моль; (9.4)
+СН2СНСН2СН3 СН3СНСН2СН3 + 79 кДж/моль; (9.4)

 + + +
+ + +
 СН3 СН3
СН3 СН3
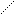 СН3СН2СНСН2ССН2СН3 СН2СН=СН2 +С(СН3)2СН2 – 21 кДж/моль (9.7)
СН3СН2СНСН2ССН2СН3 СН2СН=СН2 +С(СН3)2СН2 – 21 кДж/моль (9.7)


 (СН3)3С + + +. (9.9)
(СН3)3С + + +. (9.9) (СН3)3С
(СН3)3С СН2СНСН3+СН3СН=СН2 СН2=ССН3+СН3СНСН2СН3 +73 кДж/моль. (9.10)
СН2СНСН3+СН3СН=СН2 СН2=ССН3+СН3СНСН2СН3 +73 кДж/моль. (9.10) СН3СНСН2СН3+СН3СН(СН3)2 СН3(СН2)2СН3+ С(СН3)3+41кДж/моль.
СН3СНСН2СН3+СН3СН(СН3)2 СН3(СН2)2СН3+ С(СН3)3+41кДж/моль. RH + L R+ + LH- (9.14)
RH + L R+ + LH- (9.14)
 изомеризация
изомеризация СН3(СН2)4СН+СН3 СН3 – СН = СН2 + СН3(СН2)2СН2
СН3(СН2)4СН+СН3 СН3 – СН = СН2 + СН3(СН2)2СН2
 СН3(СН2)3СН+СН2СН3 СН3 –СН2 - СН = СН2 + СН3СН2СН2
СН3(СН2)3СН+СН2СН3 СН3 –СН2 - СН = СН2 + СН3СН2СН2
 СН3(СН2)3С+(СН3)2 СН2 = С(СН3)2 + СН3СН2СН2
СН3(СН2)3С+(СН3)2 СН2 = С(СН3)2 + СН3СН2СН2
 СН3
СН3

 Н3С
Н3С
 СН3
СН3



 СН3
СН3
СН3
СН3

 R +
R +

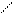
 + CH2 = C – (CH2)3CH2. (9.15)
+ CH2 = C – (CH2)3CH2. (9.15) CH2 = C – (CH2)3CH2 CH2 = C – СН(CH2)2CH3. (9.16)
CH2 = C – (CH2)3CH2 CH2 = C – СН(CH2)2CH3. (9.16)

 R R
R R
 CH2 =C(R)–СН-(CH2)2CH3 + R//CH = CH2
CH2 =C(R)–СН-(CH2)2CH3 + R//CH = CH2 +
+ CH2 =C(R)СН =CHCH2CH3 + R//CHCH3. (9.18)
CH2 =C(R)СН =CHCH2CH3 + R//CHCH3. (9.18)


 С – H
С – H  (9.19)
(9.19)









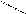 НС
НС

 С – Н + + (9.21) Н+ СН3 СН3
С – Н + + (9.21) Н+ СН3 СН3











 CH2CH2R CH2 = CHR + Н+. (9.25)
CH2CH2R CH2 = CHR + Н+. (9.25)