
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Количественные и структурные аномалии аутосом
Синдром Дауна (болезнь Дауна) Впервые была описана в 1866г английским педиатром Л. Дауном, но только в 1959г французским генетиком, врачом Дж. Леженом и соавторами было доказано, что это заболевание хромосомной природы, а именно – трисомия по 21 хромосоме. В настоящее время болезнь изучена достаточно полно, т.к является самой частой формой хромосомной патологии человека (частота синдрома среди новорожденных составляет 1: 700-800).Частота рождения больных напрямую связана с возрастом матери и отца, чем старше родители, тем чаще рождение больного ребенка. Цитологически доказано, что 20 % случаев синдрома Дауна имеют отцовское происхождение (аномальный сперматогенез), 80% - материнское (аномальный овогенез). После 35 лет у женщин существенно возрастает вероятность рождения детей с синдромом Дауна, а в возрасте 49 лет она составляет 1: 12.
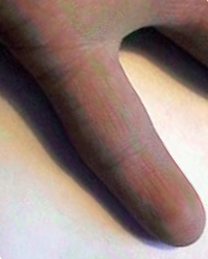
Самый распространенный вариант – трисомный. Кариотип больного- 47 хромосом, лишняя № 21(рис.7). Соотношение полов 1: 1. Клиническая картина. Дети с синдромом Дауна рождаются в срок, но с умеренно выраженной пренатальной гипоплазией (на 8-10% ниже средних величин). Череп брахицефалической формы с укорочением передне-заднего размера и уплощением затылка. Роднички шире, чем у здоровых детей и закрываются позже. Более чем у 50% детей имеется третий родничок. Избыток кожи на затылке отмечается у 80% больных. Разрез глаз «монголоидный», часто у больных выявляется вертикальная кожная складка у внутренних уголков глаз – эпикант. По краю радужек глаз обычно можно увидеть мелкие белесоватые пятна (пятна Брушфильда). Переносица широкая и вдавленная. Ушные раковины уменьшены и деформированы. У 60% детей с синдромом Дауна рот открыт, язык крупный, складчатый и виден между губами. Губы у большинства детей широкие, сухие, потрескавшиеся. Часто встречаются деформация и деструкция зубов. Руки короткие и широкие, мизинец укорочен, на ступнях обычно сандалевидная щель (увеличение расстояния между I и II пальцами).
Мышечный тонус при этом заболевании снижен. Поэтому новорожденные дети с синдромом Дауна обычно лежат в кроватке, раскинув ручки и ножки. При синдроме Дауна часто обнаруживаются разнообразные пороки внутренних органов. Около 40% больных имеют врожденные пороки сердца, несколько реже выявляются аномалии желудочно-кишечного тракта. Возможны также нарушения формирования и других органов.Синдром сопровождается расстройствами эндокринных желез и нарушением обмена веществ. Умственное развитие больных отстает (дебильность, имбецильность, идиотия). Отмечается задержка формирования моторных навыков и речи. Дети позже начитают ходить и говорить. У них резко нарушено абстрактное мышление. Им легче осваивать навыки, связанные с физическими движениями, чем речевые. Больные часто послушные, ласковые, внимательные, терпеливые при обучении. Диагностика. Цитогенетическое исследование хромосом. Лечение. Специфического лечения нет. Реабилитация больных с синдромом Дауна должна быть комплексной. Она включает массаж, гимнастику, лекарственную терапию. Питание должно быть полноценным. Большое значение придают воспитанию, занятиям с логопедом. Необходим внимательный уход за больным ребенком, защита его от простуды, инфекций. Прогноз. Умственное и физическое развитие детей протекает с задержкой. Некоторые больные могут учиться во вспомогательной школе. Синдром сочетается с острым лейкозом. Эти дети часто болеют пневмониями, тяжело переносят детские инфекции. Врожденные пороки внутренних органов и недостаточность иммунной системы часто приводят к летальному исходу в первые 5 лет жизни. Средняя продолжительность жизни больных 20-30лет, однако, известны случаи, когда больные доживают до глубокой старости. Синдром Патау (синдром трисомии 13) Генетическая природа синдрома была расшифрована в 1960г американским генетиком К.Патау и соавторами. Кариотип – 47 хромосом, трисомия № 13, чаще рождаются у матерей старшего возраста. Частота заболевания 1: 6000, соотношение полов 1: 1. Простая трисомия встречается в 80-85% случаев, примерно 15% случаев синдрома Патау являются результатом транслокации 13-й хромосомы на какую-нибудь из хромосом группы D. Значительно реже встречаются другие цитологические варианты этого синдрома: мозаицизм, изохромосомы и т.д. Беременность таким плодом обычно осложняется угрозой выкидыша и многоводием. Хромосома №13 значительно крупнее 21-й хромосомы и соответственно ее трисомия вызывает значительно более тяжелые нарушения в организме ребенка. Клиническая картина. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25%-30% ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38, 3 нед). Для синдрома Патау характерны врожденные пороки развития головного мозга и лица. Окружность черепа обычно уменьшена; лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные. Наиболее характерными пороками развития синдрома Патау являются расщелины верхней губы и неба, дополнительные пальцы на кистях и стопах (полидактилия), чаще это шестипалость. Дерматоглифика: наиболее часто описывают «обезьянью складку». Для детей с синдромом Патау характерны пороки развития внутренних органов. В 80% случаев у них отмечаются дефекты перегородок сердца. Половина детей с этим заболеванием имеют незавершенный поворот кишечника, изменение структуры поджелудочной железы и печени. Пороки почек при синдроме Патау встречаются у большинства больных, возможно нарушение развития мочеточников. Половые органы поражаются более чем в 50% случаев: у девочек наблюдается удвоение матки и влагалища, у мальчиков – гипоплазия полового члена и крипторхизм (неопущение яичек). Пороки развития органов зрения – анофтальмия (отсутствие глазных яблок), микрофтальмия (недоразвитие глаз), помутнение хрусталика, дисплазия сетчатки встречается более чем у 70% больных. Центральная нервная система поражается в 100% случаев. Масса мозга уменьшена, часто отсутствует передний мозг, недоразвитие мозжечка, иногда мозг не разделен на полушария. Диагностика. Цитогенетическое исследование хромосом. Лечение. Симптоматическое. Уход за такими пациентами направлен на облегчение их состояния, предупреждение инфекционных осложнений. Прогноз. В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни (95% - до 1 года). В возрасте старше трех лет остаются в живых единицы. Все дети с синдромом Патау имеют тяжелую умственную отсталость (глубокая идиотия). Синдром Эдвардса (синдром трисомии 18) Генетическая природа синдрома была расшифрована в 1960г Д.Эдвардсом и соавторами. Кариотип – 47 хромосом, трисомия № 18. Частота заболевания 1: 7000, соотношение полов 3: 1, причины преобладания патологии у девочек пока не ясны. Беременность при синдроме Эдвардса осложняется угрозой прерывания и многоводием. Клиническая картина. При рождении дети имеют низкую массу тела, в среднем 2180г. Синдром характеризуется задержкой роста, ярко выражен черепно-лицевой дизморфоз: череп долихоцефалической формы с выступающим затылком, лобные кости в области родничка запавшие. Низкое расположение ушей, маленькая нижняя челюсть. Чаще всего регистрируются дефекты развития конечностей, недоразвитие больших пальцев рук и лучевых костей, неправильно сформированные стопы с выступающей пяткой и провисанием свода – «стопа-качалка». Иногда (5% случаев) обнаруживаются спинномозговые грыжи и расщелины верхней губы. У детей с синдромом Эдвардса отмечается флексорное положение кистей рук. При этом III и IVпальцы прижаты к ладони и частично перекрыты II и V пальцами. Дерматоглифика характеризуется увеличением дуг на пальцах. Из пороков внутренних органов наиболее постоянны пороки сердца и крупных сосудов (90%) – нарушения межжелудочковой перегородки, клапанные пороки. Достаточно часто выявляются пороки развития желудочно-кишечного тракта: атрезия (отсутствие отверстия) пищевода, незавершенный поворот кишечника и т.д. Также отмечаются недоразвитие легких, сращение почек, удвоение мочеточников, крипторхизм у мальчиков. Диагностика. Цитогенетическое исследование хромосом. Лечение. Симптоматическое. Уход за больными в основном заключается в предупреждении инфекционных осложнений. Прогноз. Продолжительность жизни у детей с синдромом Эдвардса резко снижена - 30% новорожденных умирают в первый месяц, 50% - на втором, до года доживают немногим больше 10%. Синдром «кошачьего крика» Синдром структурной аномалия одной из хромосомы № 5 (укорочение на одну треть короткого плеча - делеция) – впервые описал в 1963г Дж.Лежен.
Клиническая картина: Масса тела ребенка с этим синдромом обычно несколько ниже. В период новорожденности плач напоминает крик кошки, что связано с аномалиями развития гортани – сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой, уменьшением надгортанника. Дети плохо растут, отстают в психическом развитии. Часто у этих детей диагностируется микроцефалия, лунообразное лицо с гипертелоризмом, эпикант, страбизм, низко расположенные уши, короткая шея, мышечная гипотония. Врожденные пороки развития внутренних органов встречаются редко, наиболее часто поражается сердце (дефекты межжелудочковой и межпредсердной перегородок). Все больные имеют тяжелую степень умственной недостаточности. Диагностика. Цитогенетическое исследование хромосом. Лечение. Симптоматическое. Прогноз. Большинство детей погибают в первые годы, около 10% больных достигают 10-летнего возраста. Аномалии половых хромосом Это группа хромосомных болезней, представленная различными комбинациями дополнительных Х- и У-хромосом, а в случаях мозаицизма - комбинациями разных клонов. Синдром трипло-Х (синдром трисомии Х) Впервые цитогенетика синдрома была описана в 1959г. А.Джекобсом. Кариотип 47 хромосом, трисомия № 23 – ХХХ, рождаются девочки. Частота заболевания 1: 1000 новорожденных девочек. Как правило, физическое и психическое развитие у женщин с этим синдромом не имеет отклонений от нормы. Это объясняется тем, что у них инактивируются две Х-хромосомы, а одна продолжает функционировать, как у нормальных женщин. Как правило, у женщин с кариотипом ХХХ не отмечается отклонений в половом развитии, такие индивиды имеют нормальную плодовитость, хотя риск хромосомных нарушений у потомства и спонтанных абортов повышен. Интеллектуальное развитие нормальное или в пределах нижней границы нормы. Лишь у некоторых женщин с трипло-Х отмечаются нарушения репродуктивной функции (вторичная аменорея, дисменорея, ранняя менопауза и др.). При тетрасомия Х наблюдается высокий рост, телосложение по мужскому типу, отклонения в умственном развитии, черепно-лицевые дизморфии, аномалии зубов, скелета и половых органов. Синдром Клайнфельтера Впервые описан в 1942г. Г.Ф.Клайнфельтером, а в 1959г. П.Джекобс и Дж.Стронг подтвердили хромосомную этиологию данного заболевания. Кариотип 47 хромосом, трисомия № 23 – ХХУ, рождаются мальчики. Частота 1: 500-700 новорожденных мальчиков. Редко встречаются индивидуумы с полисомией, имеющие 3 или 4 Х-хромосомы в кариотипе (48, ХХХУ; 49, ХХХХУ). Клиническая картина. В период новорожденности заподозрить этот диагноз невозможно. У грудных детей иногда определяются маленькие размеры яичек. Характерная клиническая картина формируется у мальчиков в пубертатном периоде. Классическими проявлениями этого заболевания считаются высокий рост, непропорционально длинные конечности, нарушение сперматогенеза, уменьшение яичек. Вторичные половые признаки выражены слабо: рост волос на лице скудный, голос сохраняет высокий тембр. Телосложение несколько феминизировано с отложением жира на бедрах и в нижней части живота. У 25% пациентов обнаруживается увеличение молочных желез - гинекомастия. У некоторых больных выявляется катаракта, снижение слуха, врожденные пороки сердца, варикозное расширение вен. Умственная отсталость отмечается в 25-50% случаев. Степень умственной отсталости колеблется от пограничных состояний до дебильности различной тяжести. Такие больные вялы, апатичны, безынициативны; им свойственны неустойчивость внимания, повышенная утомляемость и отвлекаемость, снижение работоспособности. Мужчины, страдающие синдромом Клайнфельтера бесплодны. Диагностика. Цитогенетическое исследование хромосом. Лечение. Окончательно не разработано. Больным с нормальным интеллектом проводят в пубертатный период лечение андрогенами, которые корректируют вторичные половые признаки. Однако подобная терапия не приводит к восстановлению сперматогенеза. Наиболее эффективным методом лечения гинекомастии является хирургический. Синдром полисомии У-хромосомы Кариотип 47 хромосом, трисомия № 23 – ХУУ. Частота этой патологии составляет 1: 1000 новорожденных мальчиков. Описаны индивидуумы с кариотипом 48, № 23 – ХУУУ и 49, № 23 – ХУУУУ. Клиническая картина. Большинство мужчин с таким набором хромосом не отличаются от нормальных индивидов по физическому и умственному развитию, имеют рост выше среднего (более 190см). Заметных отклонений ни в половом развитии, ни в гормональном статусе, ни в плодовитости у большинства ХУУ – индивидов нет. У таких лиц отмечаются некоторые особенности поведения: они склонны к агрессии и даже криминальным поступкам. Синдром Шерешевского-Тернера (ХО). Впервые клиническую картину синдрома описал Шерешевский в 1925г., в 1938г.Тернер дал полное описание этого заболевания, которое получило название синдрома Шерешевского-Тернера. В 1959г. Ч.Фордом было установлено, что кариотип больных – 45 хромосом, моносомия № 23 – ХО. Частота синдрома Шерешевского-Тернера среди новорожденных девочек составляет 1: 2000 – 1: 5000. Лишь 50% пациенток с синдромом Шерешевского-Тернера имеют простую полную моносомию (45, ХО). Остальные случаи –это мозаицизм (30-40%) и наиболее редкие варианты делеций, изохромосом, кольцевых хромосом. Клиническая картина. Весьма разнообразна. Наиболее частыми симптомами являются низкий рост, половой инфантилизм (наружные половые органы недоразвиты, большие и малые половые губы гипопластичны, отверстие мочеиспускательного канала расположено низко, влагалище узкое, длинное, матка недоразвита, уменьшена в размерах), вторичные половые признаки не развиты или развиты очень слабо. Молочные железы отсутствуют, соски недоразвиты, втянуты, широко расставлены друг от друга (гипертелоризм сосков); ареолы узкие, непигментированы.
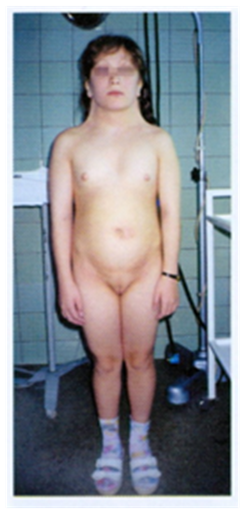 Грудная клетка расширена «щитовидная». Уши деформированы, низко расположены. Грудная клетка расширена «щитовидная». Уши деформированы, низко расположены.
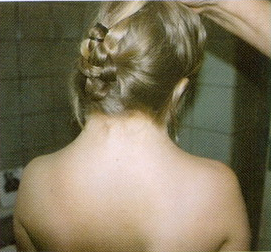
Шея короткая, широкая, у части больных встречается вертикальная кожная складка, идущая от затылка к надплечьям - шейный птеригиум (рис.11). У большинства больных наблюдают высокое, «готическое» небо. На коже часто расположено множество родинок. Нередки пороки сердца, аномалии почек. Интеллект нарушается редко. Сзади отмечается низкий рост волос (рис12). Диагностика. Цитогенетическое исследование хромосом. Лечение.
При необходимости оперативно исправляются врожденные пороки развития и косметические нарушения (удаление крыловидных складок и т.п). Генные болезни Генные болезни (ферментопатии) – это разнообразная по клинической картине группа заболеваний, обусловленная мутациями единичных генов. Начало патогенеза любой генной болезни связано с первичным эффектом мутантного аллеля.Патологический процесс, возникающий в результате мутации единичного гена, проявляется одновременно на молекулярном, клеточном и органном уровнях у любого индивида. Аутосомно-доминантные заболевания Синдром Марфана Синдром Марфана – типичная аутосомно-доминантная болезнь. Впервые описана в в1886г. В.Марфаном. Этиологическим фактором синдрома является мутация в гене, ответственном за синтез белка соединительнотканных волокон фибриллина (локализация в хромосоме 15q), приводит к блоку его синтеза, соответственно соединительная ткань обладает повышенной растяжимостью. Встречается с частотой 1: 10000-1: 15000. Мальчики и девочки поражаются одинаково часто. Клинический полиморфизм по степени выраженности симптомов очень выражен: от легких форм, трудно отличимых от нормы, до инвалидизирующего течения. Больные с синдромом Марфана имеют характерный внешний вид: они отличаются высоким ростом, астеническим телосложением, количество подкожно-жировой клетчатки у них снижено. Конечности удлинены преимущественно за счет дистальных отделов, размах рук превышает длину тела (в норме эти показатели совпадают). Отмечается арахнодактилия - длинные, тонкие пальцы на кистях и стопах. Часто наблюдается «симптом большого пальца», при котором длинный 1-й палец кисти в поперечном положении достигает ульнарного края ладони и «симптом запястья», когда при охватывании 1-м и 5-м пальцами запястья другой руки они обязательно перекрываются. Со стороны сердечно-сосудистой системы: аортальная регургитация, аневризма восходящей части аорты, пролапс митрального клапана. Со стороны органов зрения: подвывихи и вывихи хрусталиков, миопия, отслоение сетчатки, уплощение роговицы. У половины больных отмечаются паховые, диафрагмальные, пупочные и бедренные грыжи. Изредка может наблюдаться поликистоз почек, понижение слуха, глухота. Психическое и умственное развитие больных не отличается от нормы. Более чем у половины больных отмечается деформация грудной клетки - воронкообразная, килевидная «куриная», искривление позвоночника - сколиоз, грудной лордоз, гиперкифоз, высокое арковидное небо. Отмечается гиперподвижность суставов, клинодактилия мизинцев, сандалевидная щель. Прогноз жизни и здоровья определяется, прежде всего состоянием сердечно-сосудистой системы. Чаще смерть наступает от расслаивающейся аневризмы аорты. Средняя продолжительность жизни больных около 27 лет, хотя часть больных доживает до глубокой старости. При ведении беременных с синдромом Марфана необходимо помнить о возможности расслоения аневризмы аорты и последующего ее разрыва. Эти осложнения обычно возникают на поздних стадиях беременности. Лечение в основном симптоматическое. Оперативное – осторожно, в связи со слабыми аутоиммунными возможностями. Нейрофиброматоз (болезнь Реклингхаузена) Это тяжелая многосистемная болезнь с аутосомно-доминантным типом наследования. Наиболее тяжело поражается нервная система. Термин «нейрофиброматоз» (НФ) охватывает по меньшей мере две болезни НФ-1 (периферический НФ) и НФ-2 (центральный НФ). Периферическийнейрофиброматоз – одно из наиболее часто встречающихся моногенных заболеваний. В настоящее время подробно изучена его генетика и клиническая картина. ГенНФ-1 полностью расшифрован, в нем обнаружено более 100 мутаций, расположен он на 17 хромосоме. Заболевание проявляется с рождения или в первом десятилетии жизни образованием на коже светло-коричневых пигментных пятен типа «кофе с молоком», число и размер которых постепенно нарастает. Форма пятен, как правило, овальная, располагаются обычно они на закрытых участках кожи – на груди, спине, животе. С возрастом у больных на коже появляются мелкие опухоли (нейрофибромы) – от единичных до нескольких сотен. Нейрофибромы представляют собой мягкие узелки, при надавливании как бы проваливающиеся в кожу – симптом «кнопки звонка». Подкожные узелки располагаются по ходу нервных стволов (округлые бусинки диаметром 1-2 см, подвижные, не прикрепленные к коже). Нейрофибромы могут возникать в любом участке тела, захватывая кожные нервы. Часто они располагаются по ходу нервных стволов. Иногда захватывают крупные нервы и нервные сплетения (плексиформные нейрофибромы). У одного больного могут быть тысячи нейрофибром, а масса их достигает 15 кг и более, если своевременно не сделано их иссечение. У детей таких нейрофибром мало, их число увеличивается с возрастом, особенно при беременности. Пятна типа веснушек в подмышечных и паховых складках, пятнистая гиперпигментация кожи верхней части груди и промежности относятся также к частым симптомам этого заболевания. В ряде случаев, редко в детском возрасте возможно озлокачествление опухолей. Лечение оперативное. Показаниями для него являются резкая болезненность или изъязвление опухоли, затруднение движений, сдавление или смещение жизненно важных органов. В некоторых случаях к нему прибегают с косметической целью. Поскольку поражения множественные, удаление всех измененных тканей, как правило, не представляется возможным. Оперативное удаление одного из узлов в ряде случаев может привести к прогрессированию процесса с резким увеличением размеров других узлов. Во избежание прогрессирования заболевания рекомендуется избегать случайной травматизации опухолевых узлов, больные не должны подвергаться чрезмерной инсоляции и переутомляться. Ахондроплазия (врожденная хондродистрофия) Врожденная наследственная болезнь, характеризующаяся нарушением энхондрального роста костей. В результате беспорядочного расположения клеток росткового хряща нарушается процесс окостенения, и рост костей в длину задерживается. Заболевание проявляется карликовостью, короткими конечностями при обычной длине туловища, деформацией нижних конечностей и позвоночника, относительной макроцефалией. Ахондроплазия развивается вследствии мутации гена рецептора типа 3 фактора роста фибробластов, наследуется по аутосомно-доминантному типу. Уже при рождении у ребенка увеличена мозговая часть черепа и выпуклый лоб, что обусловлено аномалией развития хрящевой основы черепа. Желудочки значительно увеличены, но давление внутри желудочков, как правило, нормальное. Вследствие выраженного нарушения развития костей основания черепа у больных выдающиеся вперед лобные кости, седловидный нос, прогнатия (выступание верхней челюсти вперед). Поражается весь скелет, но особенно выраженные изменения отмечаются в проксимальных отделах конечностей. Все трубчатые кости утолщены, изогнуты, бугристы. Галифеобразно искривлены и несколько скручены внутрь бедренные кости в дистальной трети. Наблюдается некоторый изгиб плечевой кости. Кисти и стопы маленькие, широкие. Пальцы на кистях короткие (брахидактилия) расположены веерообразно или в виде трезубца вследствие сближения III и IV пальцев. Наблюдается изодактилия – одинаковая длина пальцев. Дети, больные ахондроплазией, отстают в физическом развитии, поздно начинают держать головку (после 3-4 месяцев) и сидеть (после 8-9 месяцев), а ходить начинают к 1, 5-2 годам. Уже на первом году жизни у многих больных появляется кифоз в поясничном отделе позвоночника. По мере роста ребенка укорочение костей становится более заметным. Лечение в раннем возрасте направлено на профилактику деформаций нижних конечностей, спины и живота. Хирургическое лечение проводят при развившихся деформациях нижних конечностей, а также с целью увеличения роста. Аутосомно-рецессивные заболевания Нарушение обмена аминокислот: фенилкетонурия Фенилкетонурия (ФКУ, фенилпировиноградная олигофрения, болезнь Феллинга) – это одна из самых частых форм наследственных дефектов обмена аминокислот. Клинически ФКУ в самостоятельную форму выделена А.Феллингом в 1934г. Частота в европейских популяциях составляет 1: 10000 новорожденных, а частота гетерозигот 1: 100. Болезнь поражает лиц обоего пола, но девочек несколько чаще. Наследование – аутосомно-рецессивное, мутантный ген картирован, локализован в длинном плече 12-й хромосомы. Больные дети нередко рождаются у здоровых родителей, которые являются гетерозиготными носителями мутантного гена. Родственные браки резко повышают возможность появления ребенка, гомозиготного по гену ФКУ. В основе заболевания лежит дефицит фермента фенилаланин-4-гидроксилазы, который контролирует превращение аминокислоты фенилаланин в тирозин. В результате энзимного блока концентрация фенилаланина в организме больного увеличивается в десятки раз. Часть его выводится с мочой, а остальное количество превращается в фенилпировиноградную кислоту и другие фенилкетоновые производные, которые в норме присутствуют в организме, но в очень небольших концентрациях. Повышение концентрации этих токсических веществ ведет к нарушению формирования миелиновой оболочки вокруг аксонов в центральной нервной системе. Клиническая картина. Новорожденный с ФКУ внешне ничем не отличается от здоровых малышей. Но с началом кормления в организм начинает поступать фенилаланин с молоком матери или с искусственным детским питанием и возникают первые симптомы заболевания. В первые месяцы жизни ребенок может быть беспокойным или, наоборот, вялым, сонливым. Отмечается побледнение кожи, волос, радужной оболочки (результат снижения уровня тирозина). От больного ребенка исходит своеобразный «мышиный» запах. К полугодию выявляется задержка психомоторного развития. Физическое развитие нарушено в меньшей степени, рост обычно нормальный, но может быть и снижен. Отмечается некоторое уменьшение размеров черепа, позднее прорезывание зубов. Моторное развитие задержано, дети с запозданием начинают сидеть, ходить. В дальнейшем поза больного и походка очень своеобразны. Такие дети стоят, широко расставив ноги, согнутые в коленях и тазобедренных суставах, опустив плечи и голову. При ходьбе делают маленькие шаги и покачиваются. Больные сидят обычно в «положении портного» - поджав ноги. У детей после трех лет клиническая картина характеризуется умственной отсталостью (в 95% случаев это имбецильность или идиотия), нарушением поведения (чаще возбудимость, расторможенность, психотические расстройства), судорожным синдромом. Ранняя диагностика ФКУ (неонатальный скрининг) и профилактическое лечение (специфическая диета) предупреждает развитие клинической картины болезни. Лечение. Для достижения оптимального эффекта дети с ФКУ должны переводиться на специальную диету на 2 – 3-й неделе жизни. Диетотерапия (резкое ограничение в пище фенилаланина) проводится постоянно до пубертатного возраста под контролем уровня фенилаланина в крови. Аминокислота фенилаланин входит в структуру большинства белков, поэтому из рациона больного исключаются все высокобелковые продукты: мясо, рыба, творог, яичный белок, хлебо-булочные изделия и др. Фактически питание ребенка ограничивается небольшим количеством овощей и фруктов, которые содержат относительно мало белка. В пищу детей вводят специальные низкобелковые продукты (особый хлеб, вермишель и др.), приготовленные на основе кукурузного крахмала. Для компенсации необходимой дозы белка в питании растущего организма используют искусственно созданные белковые продукты или аминокислотные смеси, не содержащие фенилаланин: для грудных детей – «Лофеналак», «Аденилак», «Фенил-40»; для детей старше года – «Фенил-фри», «Тетрафен», «Фенил-100» и др. Раннее введение диеты (на 1-м месяце жизни) и ее регулярное соблюдение обеспечивают практически нормальное развитие ребенка. Нарушение обмена углеводов Галактоземия Заболевание, впервые было описано в 1908г., связано с невозможностью использования организмом галактозы и проявляющееся в виде тяжелого поражения печени, нервной системы, глаз и других органов. Ген галактоземии локализован на коротком плече 9-й хромосомы. Наследование – аутосомно-рецессивное. Частота заболевания 1: 20000 новорожденных. Причиной классической формы галактоземии является дефицит фермента галактозо-1-фосфоуридилтрансферазы, который приводит к накоплению в тканях больного ребенка галактозо-1-фосфата. Галактоза – основной фермент молока, в том числе и женского. Поэтому патологические изменения возникают с первых дней жизни ребенка, как только он начинает вскармливаться молоком. Клиническая картина. Упорная обильная рвота, реже понос возникают вскоре после кормления ребенка молоком. Быстро развивается гипотрофия. Дети отказываются от еды. Появляется желтушность кожи, которая не исчезает и после периода новорожденности, увеличивается печень, часто до больших размеров (цирроз печени). Селезенка увеличена незначительно. При приеме молочной пищи у ребенка регистрируется низкий уровень глюкозы в крови. В первые месяцы жизни ребенка формируется помутнение хрусталиков глаз (катаракта), нарушаются функции почек. Постепенно становится заметной задержка умственного и физического развития, возможно возникновение судорог, даже смерть на фоне очень низкого уровня глюкозы в крови или цирроза печени. Главным в лечении галактоземии является назначение специальной диеты, не содержащей продуктов с галактозой. С первых дней жизни ребенок должен быть переведен на безмолочное вскармливание. В качестве заменителей молока – смеси, в состав которых обычно входит соевое или миндальное молоко, казеиновые гидролизаты с удаленной лактозой (которая при нормальном обмене превращается в галактозу и глюкозу), яйца, растительные масла и другие продукты. Каши готовят на овощных и мясных отварах. В настоящее время известны и другие типы галактоземии, которые не сопровождаются тяжелым нарушением состояния здоровья. Так, при атипичных вариантах заболевания, связанных с дефицитом галактокиназы и уридиндифосфогалактозо-4-эпимеразы клинические проявления обычно отсутствуют. Ранняя диагностика галактоземии (неонатальный скрининг) и раннее исключение из пищи больных галактозы предупреждает развитие клинической картины болезни. При тяжелой форме заболевания и отсутствии лечения смерть наступает в течение нескольких месяцев в результате печеночной или сердечно-сосудистой недостаточности. Нарушение обмена липидов Наследственные дефекты обмена липидов включают гиперлипидемии и внутриклеточные липидозы (гликолипидозы). Гиперлипидемия сопровождается повышением уровня липидов в плазме крови, в которой они содержатся как в комплексе с альбумином, или в виде эмульсии, так и образуя сложные химические структуры – липопротеиды. Липопротеиды обеспечивают транспортировку холестерина и триглицеридов к органам. Причиной наследственных гиперлипидемий обычно является нарушение обмена липопротеидов. Избыток липидов в крови приводит в первую очередь к поражению органов сердечно-сосудистой системы. Иногда тяжелые атеросклеротические изменения развиваются уже в детском возрасте. У больных часто обнаруживаются ксантомы – участки подкожного отложения холестерина. Внешне это опухолевидные образования разного размера, желтоватой или желтовато-оранжевой окраски, обычно расположенные в области суставов, на веках. Кроме того, при этих заболеваниях часто развиваются панкреатит и ишемическая болезнь сердца, увеличиваются размеры печени, нарушается кровоток в сосудах конечностей. Такие пациенты могут погибнуть в молодом возрасте от инфаркта миокарда. Самым распространенным заболеванием из группы гиперлипидемий является семейная гиперхолестериномия. В основе заболевания – 4 класса мутаций генов рецепторов липопротеидов. При гомозиготности по этим мутациям уже в детском возрасте отмечается высокий уровень холестерина в крови, множество ксантом. К 14 годам у больных развивается атеросклероз аорты и коронарных сосудов. Большинство таких больных погибает до 30 лет от инфаркта миокарда. Гомозиготы по данной патологии встречаются с частотой 1: 250000 человек. У гетерозиготных носителей этих мутантных генов признаки ишемической болезни сердца и ксантомы возникают к 40-50 годам. Лечение гиперлипидемии включает специальную диету, ограничивающую поступление в организм жиров, в первую очередь животных, а также медикаментозные препараты, снижающие уровень холестерина и липопротеидов крови и ограничивающих их всасывание в кишечнике. Внутриклеточные липидозы характеризуются накоплением в клетках сфинголипидов, которые являются составной частью вещества мозга. Кроме того, они входят в качестве компонентов в различные мембраны организма. Заболевания этой группы встречаются достаточно редко и в основном наследуются по аутосомно-рецессивному типу, за исключением синдрома Фарби, ферментный дефект при котором связан с Х-хромосомой. Рассмотрим некоторые заболевания этой группы: болезнь Гоше, болезнь Тея-Сакса, болезнь Нимана-Пика. Болезнь Гоше. В основе заболевания лежит утрата активности фермента глюкоцереброзидазы, приводящая к накоплению в клеткахнакоплению в клетках ретикулоэндотелиальной системы, особенно в печени, селезенке и костном мозге глюкоцереброзида. Заболевание наследуетсяпо аутосомно-рецессивному типу. Основными симптомами заболевания являются наклонность к носовым кровотечениям, кожным гемморагическим высыпаниям, обильным маточным кровотечениям. Характерным признаком болезни Гоше считается увеличение печени и селезенки, последняя достигает огромных размеров, занимая почти весь живот. Не менее важным диагностическим признаком является изменение костной системы. Возникают боли в трубчатых костях, усиливающиеся при движении. Череп обычно не поражается. Важным симптомом заболевания (встречается примерно у половины больных) – своеобразная коричневая окраска кистей рук, лица, носящая пятнистый или диффузный характер. |
Последнее изменение этой страницы: 2017-05-05; Просмотров: 3050; Нарушение авторского права страницы

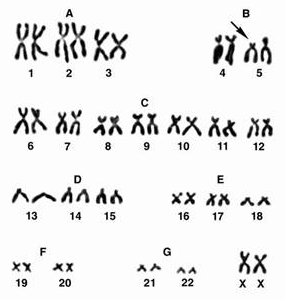

 Женщины, страдающие синдромом Шерешевского-Тернера бесплодны.
Женщины, страдающие синдромом Шерешевского-Тернера бесплодны.