
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Гидродинамическая хроматография
Гидродинамическая хроматография (ГДХ) жидкостная хроматография, в которой роль неподвижной фазы играют стенки колонки (канала) и разделение смеси макромолекул или частиц размером от нескольких десятков нанометров до нескольких микрометров происходит вследствие различия скоростей протекания подвижной фазы вдоль оси канала и у его стенок, а также за счет распределения разделяемых частиц по сечению канала в соответствии с их размером. Колонка заполняется твердыми непористыми сферическими частицами из стекла, пластика или ионообменной смолы размером от 10 до 50 мкм в зависимости от эффективности колонки. Вместо колонки может применяться полый капилляр. В последнем случае говорят о капиллярном варианте ГДХ. Для целей ГДХ может быть использован типовой жидкостной хроматограф. Возможно, первым исследователем, наблюдавшим сегрегацию частиц в капилляре, был Пуазейль, который заметил наличие возле стенки трубки области, свободной от частиц красителя. Разработал метод ГДХ Смолл в 1974 г. К своему открытию Смолл пришел в опытах, в которых он пытался распространить методику эксклюзионной хроматографии на определение размеров коллоидных частиц на уровне 1-2 мкм. Из-за чрезвычайно медленной броуновской диффузии таких частиц в жидкости их массообмен в пористой среде затруднен, что приводило к интенсивному размыванию пиков и низкому разрешению. Как альтернатива гелъ-хроматографического варианта по аналогии с сорбционио-десорбционным механизмом разделения компонентов на молекулярном уровне был предложен гипотетический механизм разделения коллоидов на основе процессов флоккуляции − дефлоккуляции. Велся поиск условий для реализации такого механизма. В опытах по разделению частиц полистирольного латекса на слое катионита в потоке де-ионизованной воды при очень низком разрешении был обнаружен необычный эффект. Частицы большего размера элюировали перед частицами меньшего размера, что противоречило предполагаемому механизму флоккуляции – дефлоккуляции. Кроме того, выбранные хроматографические условия, особенно низкая ионная сила подвижной фазы, вообще не шособствовалк процессу флоккуляции. Проведенные оценки исключали возможность эксклюзии по размерам в слое катионига. Для объяснения наблюдаемых эффектов была привлечена капиллярная модель, согласно которой свободное пространство между заполняющими колонну непористыми частицами рассматривается как система капилляров. Известно, что для вязкой жидкости, текущей в капилляре, существует параболический профиль скорости с максимумом по оси капилляра (поток Пуазейля). Коллоидная сферическая частица (рис. 1.26.), введенная в цилиндрический канал, благодаря броуновскому движению будет совершать радиальные перемещения, перпендикулярные направлению потока, так что ее продольная составляющая скорости соответствует локальной скорости потока. Средняя скорость коллоидной частицы поэтому будет отражать профиль скорости жидкости в капилляре за одним важным ограничением. Центр частицы из-за ее конечных геометрических размеров не будет достигать прилегающих к стенкам капилляра и имеющих наименьшую скорость слоев. Следовательно, частица будет двигаться через капилляр со средней скоростью, превышающей среднюю скорость жидкости на фактор, который возрастает с увеличением отношения размера частицы к радиусу капилляра. Чем крупнее частица, тем большее время (по сравнению частицами меньшего размера), в процессе движения по капилляру она находится в слоях с большей скоростью несущей жидкости. Это обуславливает различие в средних скоростях переноса частиц разного размера и обеспечивает при достаточной длине канала их разделение. Из сказанного становится понятным, что более крупные частицы выходят из колонки первыми, а самые мелкие остаютсяся " в хвосте".
Разделение вГДХ характеризуют величиной Rf=up /um, (1.34) где иp - скорость переноса коллоида, ит - средняя скорость элюента. Последнюю оценивают по времени удерживания движущегося со средней скоростью жидкости подходящего маркера, при выборе которого необходимо, чтобы он перемещался по слою лишь в свободном объеме насадки и по своим свойствам мало отличался от элюента. Следует подчеркнуть, что обычно Rf > 1, т. е. скорость коллоидных частиц превышает среднюю скорость несущей их жидкости. Это необычное явление находится в полном противоречии с общеизвестными хроматографическими закономерностями, согласно которым и только в предельном случае для несорбирующихся компонентов Rf = 1. Рассматриваемый вариант хроматографии был назван гидродинамическим потому, что первым его исследователям поначалу казалось, что в отличие от обычных вариантов хроматографии, в основе которых лежат определенные физико-химические взаимодействия между сорбатом и неподвижной фазой, в этом методе силы, ответственные за разделение, определяются факторами, действующими исключительно в свободном объеме слоя насадки. Точнее в пространстве между зернами, доступном для гидродинамического потока, без учета внутренней пористости элементов слоя. Однако последующие экспериментальные и теоретические исследования показали, что гидродинамика потока - всего лишь один из факторов, определяющих Rf и механизм взаимодействия имеет гораздо более сложный характер. Например, на процесс разделения влияет потенциал взаимодействия коллоидных частиц со стенками капилляра. Наиболее общий характер носят дисперсионные взаимодействия Лондона - Ван-дер-Ваальса и электростатические взаимодействия двойных электрических слоев. Теория гидродинамической хроматографии проанализирована в работе. Еще одной особенностью ГДХ является то, что хроматографируемые частицы в большинстве случаев практически нерастворимы в подвижной фазе. Они находятся в виде суспензии, причем размер частиц может быть достаточным для наблюдения за ними с помощью микроскопа. Это так же противоречит одному из главных правил жидкостной хроматографии, согласно которому дня успешного проведения хроматографического процесса разделяемые вещества должны быть полностью растворимы в подвижной фазе. В качестве детектора в ГДХ обычно используют турбидиметр. В зависимости от длины волны детектор работает либо по механизму, сочетающему рассеяние с химической абсорбцией, либо только - рассеяния света. Для коллоидов с очень малыми частицами (малыми по отношению к длине волны света λ ) мутность пропорциональна шестой степени диаметра частиц d, для более крупных частиц эта зависимость менее резкая. Как следствие сигнал от малых частиц сравнительно слабый, хотя он может быть увеличен за счет использования более коротковолнового источника. Следует отметить, что при измерениях распределений частиц по размерам больший интерес представляет не абсолютный, а относительный сигнал. Для неабсорбирующнх частиц изменения λ или отношения коэффициентов преломления коллоида и среды слабо влияют на относительные показания детектора. При абсорбции относительный сигнал возрастает благодаря значительному усилению коэффициентов поглощения малых частиц. Кроме измерения мутности полимерной суспензии, фотометр должен давать информацию об оптической плотности зоны элюирования молекулярных маркеров, по которой рассчитывают среднюю скорость несущей жидкости. Оптическая плотность направляемого из колонки в детектор потока не должна превышать некогорого предела, чтобы обеспечить согласно закону Бера линейность зависимости оптической плотности от концентрации частиц. Зависимость поглощения и рассеяния света коллоидной суспензией как от размера частиц, так и от их концентрации, усложняет интерпретацию результатов. В раде случаев зависимостью or d пренебрегают. Помимо турбидиметра в ГДХ, получил распространение дифференциальный рефрактометрический детектор. По сравнению с турбодиметрией неабсорбирующих частиц показания дифференциального рефрактометра существенно меньше зависят от размера частиц. При уменьшении среднего размера частиц насадки величина Rf возрастет, т. е. разрешение улучшается. Зависит Rf не только от размера разделяемых частиц и размера частиц насадки, но и от ионной силы подвижной фазы. Увеличение ионной силы во всех случаях приводит к уменьшению Rf.Это связано с электростатическими взаимодействиями двойных электрических слоев поверхности частиц насадки и коллоидных частиц. Что касается влияния скорости подвижной фазы, то с eе повышением величина Rf сначала уменьшается, а при достижении минимума (при рабочем давлении около 8 МПа) либо остается постоянной, либо возрастает. Капиллярная гидродинамическая хроматография реализуется в капиллярах в внутренним диаметром 75–800 мкм при скорости потока подвижной фазы 0.2–20 мкл/мин. Если гель-хроматография применима при разделении на молекулярном уровне с М до нескольких млн. дальтон, область использования ГДХ с набивными колонками - частицы с размером 0.01–1.0 мкм, то капиллярный вариант ГДХ позволяет разделять коллоидные частицы размером 0.5–30 мкм. Для реализации капиллярной ГДХ можно применять стандартные хроматографы, например, фирмы " Hewlett-Packard" серии HP 1100. Этот хроматограф может быть легко трансформирован в систему дтя высокоэффективной капиллярной ЖХ с помощью процессора микропотока установленного перед колонкой. Стандартные исходные скорости потока устанавливаются равными 100 мкл/мин и при разделении потока понижаются до 2–4 мкл/мин - величин скорости потока, оптимальных для капиллярных колонок с внутренним диаметром 300 мкм. Преимуществом такого подхода является то, что для выполнения разделений методом капиллярной ГДХ можно использовать стандартную хроматографическую систему. Метод ГДХ, как и гель-хроматография, позволяет анализировать полидисперсные системы. Развитие методов калибровки и численных методов расчета на компьютере позволяют получать кривые распределения частиц по размерам даже в случае неразделенных пиков. ГДХ, как экспрессный и не деструктивный метод анализа частиц по размерам, позволяет исследовать кинетику процессов, в ходе которых частицы меняют свой эффективный гидродинамический диаметр, в частности процессов усадки полимерных частиц, набухания, агрегации. 1. 4.13. Фракционирование в поперечном поле сил Фракционирование в потоке, находящемся в поперечном физическом поле (Bid-Flow Fractionation) - жидкостная хроматография, в которой роль неподвижной фазы играют стенки колонки (канала) и разделение смеси макромолекул или частиц происходит вследствие различия скоростей протекания подвижной фазы вдоль оси канала и у его стенок, а также за счет распределения разделяемых частиц по сечению канала в соответствии с их размерами и поведением в приложенном в поперечном направлении поле (гравитационном, магнитном, температурном). Фракционирование в потоке под действием силовых полей (FFF) методологически можно рассматривать как однофазовую жидкостную хроматографию. Силовое внешнее поле, или градиент в одной фазе, заменяет собой силы разделения и адсорбции и распределяет растворенное вещество между различными областями потока. Имея дело с одной фазой, можно полностью исключить искажения в распределении фаз и межфазовую адсорбцию, которые неизбежно возникают во всех видах хроматографии при увеличении молекулярной массы. Разделение методом FFF применяют для анализа полимеров, биополимеров (белков, дипопротеи-нов, белковых агрегатов) и даже для частиц (взвесей, микроорганизмов, микрогелей в полимерных матрицах). В настоящее время исследован диапазон от 103 до 1012 дальтон. Разделение происходит на основе использования внешнего поля или градиента для избирательного распределения растворенного вещества между различными областями потока в канале. Канал обычно не имеет наполнителя, имеет равномерное сечение, характеристики его легко рассчитываются теоретически. На рис 1.27 показана наиболее предпочтительная конфигурация канала. Теоретические основы метода FFF изложены в [197]. Практически в ходе эксперимента канал работаег гак же, как колонка в процессе жидкостной хроматографии: насос создает ноток и управляет им, образец вводится в начало канала, детектор контролирует элюируемую жидкость. Только природа канала отличается от колонки и механизм разделения совершенно другой. В сущности, канал представляет собой хроматографичеекуго колонку, которая содержит лишь подвижную фазу. В этом способе разделения внешнее поле играет ту же роль, что и стационарная фаза. Действуя перпендикулярно потоку, оно вытесняет растворенное вещество в относительно медленные области потока недалеко от одной из стенок канала. Эти квазинеподвижные области около стенок канала играют ту же роль, что и неподвижная фаза в классической хроматографии, а поля, которые управляют ими, аналогичны силам притяжения, которые создаются стационарной фазой. Теоретически разделение в поле сил имеет столько же разновидностей, сколько их имеет хроматография. Большинство основных разновидностей определяется видами " полей", которые используются для создания удерживания в силовом поле разделения. Эти поля аналогичны видами стационарных фаз в хроматографии. В хроматографии в качестве стационарных фаз применяются жидкости, твердые поверхности, ионообменные смолы, пористые материалы, жидкие кристаллы и т.д. Аналогично в качестве силовых полей можно использовать тепловые градиенты, электрические поля, поля поперечного потока, гравитационные поля. Таким образом, ясно, что разделение в поле сил так же, как и хроматография, является универсальным методом, который может быть использован для работы с водными, неполярными и полярными органическими растворителями, с молекулами произвольной структуры и с частицами, а также с веществами, молекулярная масса которых меняется в очень больших пределах.
Рис. 1.27. Форма канала с параллельными стенками для разделения в потоке под действием поперечного ноля: 1 -поток растворителя, 2 -введение образца; 3 - вектор поля, 4 - к детектору; 5 - параболический профиль потока; 6 - растворенное вещество Теоретическая тарелка в методе FFF определяется по формуле: где ν - средняя скорость потока в канале; χ – коэффициент, связанный с длиной и шириной канала сложной зависимостью: D – коэффициент диффузии растворенного вещества в растворителе; ω – ширина канала. Калибровочная зависимость для однофазовой хроматографии имеет где коэффициент q может принимать значения от 1/3 до 1. Из формулы (1.36) видно, что эта зависимость позволяет согласовать требования к широкому диапазону и хорошей разрешающей способности. На рис. 1.28. приведен пример применения метода FFF для разделения пояистирольных полимеров, который подтверждает возможность получения очень широкого диапазона, но отнюдь не говорит о том, что этот диапазон является максимально возможным. Ддя разделения использовали тепловое поле. Подвижная фаза – вода. Разделение проводили в течение одного цикла, процесс программировался. При этом использовали канал длиной 0, 4 м и шириной 0, 02 м, образованный двумя пластинами, расстояние между которыми составляло 25∙ 10-5 м. Одна металлическая пластина нагревалась, другая – охлаждалась. В качестве силового поля служил температурный градиент, создающий тепловую диффузию. Программирование процесса заключалось в постепенном понижении температуры нагреваемой пластины.
Запись кривых распределения вещества при разделении в поле сил напоминает хроматограмму из гель-хроматографии, вместе с тем особые однофазовые характеристики и иные силы, управляющие разделением, создают коренные отличия в способе перемещения и разделения растворенных вещесгв.
Рис. 1.28. Разделение полистролов методом FFF. I - 4000; 2 - 20000; 3 - 51000; 4 97000; 5 - 200000; 6 - 411000; 7 860000; 8 - 1800000; 9 • 7100000 дальтон тем особые однофазовые характеристики и иные силы, управляющие разделением, создают коренные отличия в способе перемещения и разделения растворенных вещесгв. Понимание этих отличий очень важно для уяснения недостатков и преимуществ данного способа разделения, а также для определения возможности расширения диапазона молекулярных масс до очень больших величин. Поэтому уместно рассмотреть особенности метода FFR Площади поверхности для разделения в поле сил много меньше, чем ддя хроматографии. Причина заключается в том, что эта поверхность ограничена стенками канала. Более того, материал этой поверхности легко контролировать к заменять. Основным требованием к материалу канала является способность передавать воздействие поля. Обычно этому требованию удовлетворяет широкий круг материалов. Следующая особенность однофазовой хроматографии состоит в том, что при разделении под действием силового поля возможно довольно точное описание удерживания в этой системе, что позволяет заранее выбрать оптимальные условия работы. Что еще более важно, можно предварительно дать приблизительную оценку характеристикам сложной системы, не прибегая к снятию калибровочной кривой. Вещества в каждой точке спектра элюции можно непосредственно охарактеризовать с помощью некоторых физико-химических параметров, таких, как диффузия, или с помощью ряда простых физико-химических параметров. Эти параметры могут быть, в свою очередь, связаны с молекулярной массой, распределением заряда и сдруги-ми важными свойствами растворенного вещества. Почти линейная зависимость между объемом удерживания и молекулярной массой обеспечивает достаточно широкий диапазон измерений по молекулярной массе при достаточно хорошем разделении. Это обстоятельство объясняется линейным возрастанием скорости потока с ростом расстояния от стенки к центру канала, а также обратной зависимостью от молекулярной массы. Открытый и однородный канал, распределение потока в пределах относительно большого поперечного сечения, отсутствие стационарной фазы, которая разворачивает сегменты макромолекул против потока, - все эти факторы позволяют в однофазовой хроматографии избежать деструкции макромолекул при движении вдоль канала вплоть до очень высоких значений молекулярных масс. Простым изменением величины силового поля можно получить величину объема удерживания практически в разумных пределах любой желаемой величины. Такой контроль является очень быстрым и может быть осуществлен, если потребуется, непосредственно в ходе процесса. При таком виде контроля над процессом разделения можно раздвинуть пики, слишком близко сгруппированные, улучшив тем самым их разрешение. Кроме того, путем простого снижения силы поля можно более быстро злюировать пики с очень большим временем удерживания. Другим полезным качеством такого контроля является способность полностью прекратить удерживание. Благодаря этому, полностью отпадает необходимость в такой операции, обычной в гель-хроматографии, как длительная промывка колонки с целью удаления компонентов " забивших'* колонку. В однофазовой хроматографии, если надо удалить из колонки остаток, нужно просто уменьшить воздействие силового поля до нуля и дождаться прохождения через канал объема, немного большего, чем свободный объем. Силу внешнего поля в однофазовой хроматографии можно непрерывно варьировать в соответствии с любой программой. Наибольшее преимущество состоит в том, что программу можно использовать дтя расширения диапазона молекулярных масс. Так, если сила поля непрерывно уменьшается в течение цикла, то первыми вымываются макромолекулы или частицы с низкими молекулярными массами, за ними, по мере уменьшения поля, компоненты с более высокими молекулярными массами. Степень избирательности воздействия силового ноля на разделение вещества в однофазовой хроматографии пока не определена. Несомненно, что химические свойства разделяемых макромолекул или частиц будут оказывать влияние на разделение при воздействии некоторых видов полей. Важность этого эффекта требует дальнейших исследований. По мере развития новой аналитической техники и технологии метод FFF находит все большее применение в биохимии, микробиологии и химии полимеров. Одним из перспективных направлений FFF является проточное фракционирование с асимметричным электроосмотическим потоком. Этот метод позволяет фракционировать объекты в диапазоне от I нм до 100 мкм. Он превосходит по разрешающей способности проточное фракционирование в пуазейлевском потоке на порядок. Проточное фракционирование в асимметричном электроосмотическом потоке предложено В.П. Андреевым [201-203]. В отличие от классического проточного фракционирования в этом методе продольный поток – электроосмотичеекий, причем неоднородность профиля потока, необходимая для реализации метода, достигается за счет изготовления стенок канала из материалов, имеющих отличающиеся значения дзета-потенциалов. В частности, если дзета-потенциал одной из стенок канала равен нулю, то в канале будет сформирован электроосмотический поток с линейным профилем; при неравных, но имеющих один знак дзета-потенциалах стенок реализуется трапециевидный профиль, при отличии в знаках дзета-потенциалов стенок реализуется ситуация, когда жидкость двигается в одном направлении у одной из стенок и в противоположном направлении у другой стенки. Вариант с движением жидкости в противоположных направлениях у нижней и верхней стенок канала интересен для одновременною фракционирования частично плотностям и размерам. Математическое моделирование процесса проточного фракционирования в канале с асимметричным электроосмотическим потоком подтвердила высокую эффективность и разрешающую способность метода. В частности, при определенном сочетании возникающего в канале линейного асимметричного электроосмотического и пуазейлевского потоков достигается рекордная разрешающая способность, превышающая разрешающую способность классического FFF на 2 порядка. 1.4.14. Электрофорез Электрофорез электромиграционный метод разделения, смежный с жидкостной хроматографией. Процесс разделения компонентов смеси происходит в жидком растворе электролита, т.е. жидкость является непременным и важным компонентом электрофоретической системы. Растворитель (буферный раствор) представляет собой неподвижную фазу, тогда как растворенное вещество (заряженные частицы) мигрируют под действием приложенного электрического поля. Имеются гибридные электрохроматографическис методы, в которых разделение проистекает и за счет хроматографичеекого процесса, и под влиянием внешнего электрического поля. Например, незаряженные молекулы можно разделять с помощью мицеллярной электпрокинетической хроматографии. В этом случае к буферу добавляется детергент, и нейтральные молекулы распределяются между буфером и мицеллами в соответствии с их гидрофобностью. Разделение в этом методе основано на подвижности мицелл, заряженных в большинстве случаев отрицательно. Поскольку в основе разделения лежит процесс распределения в жидкофазной среде, можно с полным основанием отнести этот метод к жидкостной хроматографии. В целом методы, в которых имеется неподвижная фаза для ЖХ, а течение элюента и перенос пробы происходит только за счет электроосмотического потока, относят к электрохроматографии. В электрохроматографии зонный электрофорез чаще всего сочетается с ионообменной хроматографией. Электрофорез основан на свойстве ионизированных частиц к электрофоретической подвижности в постоянном электрическом поле. Скорость иона онределяегся в первую очередь его зарядом, размером, напряженностью поля и Уменьше площадь сечения электрсфорегической ячейки q и электропроводность χ . Если напряженность поля можно выразить формулой
то скорость идеальной сферической частицы определяется соотношением где z – заряд частицы, Е – напряженность электрического поля, r – радиус частицы, π – вязкоегь среды. Электрофоретическая подвижность имеет размерность см2/(В∙ с) и определяется соотношением Вместе с тем приведенные выше уравнения справедливы при бесконечном разбавлении, отсутствии солей и идеальной сферической структуры частицы. В реальных условиях с повышением ионной силы снижается подвижность ионов из-за накапливания ионов противоположного знака, что приводит к снижению эффективного заряда. Безусловно, имеются отклонения в электрофоретической подвижности, связанные с не сфероидальным строением частиц. Величина и обратно пропорциональна η неподвижной фазы. Вязкость уменьшается с увеличением температуры, а подвижность возрастает в 2.5–3 раза с повышением температуры на каждый градус. Понизить подвижность частицы можно, поместив жидкую среду в твердый носитель с волокнистой структурой или порошкообразный носитель в гель. В этих случаях путь частицы удлиняется, уменьшается напряженность поля. Классический вид электрофореза метод подвижной границы по Тизелиусу, дает точную информацию об электрофоретической подвижносги биополимеров (белков, пептидов), но не пригоден из-за низкого разрешения для выделения чистых компонентов анализируемой смеси. Так, если подвижность компонентов уменьшается в раду А, В, С, А то в результате электрофореза появляется зона, обогащенная компонентом А, за ней следуют зоны смесей А+B, А+В+С, A+B+C+D, B+C+D, C+D и, наконец, зона самого медленного компонента D. При разделении методом зонного электрофореза перемещение компонентов смеси продолжается до полного их разделения. В этом методе используют неподвижный носитель, по поверхности или через объем которого осуществляется миграция ионов. Носители могут иметь вид полос (например, из бумаги, колонок, дисков, тонких слоев и др.). При проведении электромиграционных процессов важно, чтобы у концетрационных зон были четкие границы. Диффузию можно ограничивать при помощи градиента плотности, вращения, камер с извилистыми каналами, капиллярного эффекта, ламинарного потока, возрастающей вязкости, если среда жидкая. В пористой среде антиконвекционную стабилизацию зон обеспечивают применением различных типов носителей с волокнистой структурой (разнообразные виды бумаги), гомогенных пленок на основе модифицированной целлюлозы, незакрепленных слоев с молекулярно-ситовыми свойствами (целлюлоза, крахмал, гранулированные гели). Кроме того, процессы проводят в различных гелях (гель-электрофорез) на основе крахмала, агарозы, полиамидов. Электрофорез реализуют под действием нолей низкого (< 1000 В) и высокого напряжения (1000–10000 В). Высоковольтный электрофорез проводят, как правило, на бумаге. Этот метод дает хорошие результаты при анализе аминокислот и других небольших молекул и непригоден для анализа макромолекул. Обнаружение разделенных компонентов на электрофорерограмме выполняют, погружая в раствор со специфичным реагентом или опрыскивая бумагу этим реагентом. При разделении белков и пептидов продуктивен метод изоэлектрофокусироваия. Разделение смеси проводят в среде, представляющей собой смесь амфотерных соединений, образующих устойчивый градиент рН в постоянном электрическом поле. Соединения с различными изоэлектрическими точками мигрируют до такого положения, в котором рН равен их изоэлектрическим точкам. Устанавливается квазистационарное состояние, миграция соединений прекращается, диффузия компенсируется фокусирующим влиянием электрического поля при градиенте рН. Гипотетическая смесь компонентов А, В, С и D вэтом случае расположится между низкомолекулярными амфотерными соединениями Мi: М1А, М2В, М3С, М4D, М5. Где Мi - добавленные промежуточные фракции с различными изоэлектрическими точками. В изотахофорезе используют два электролита - лидирующий электролит, который содержит ион с максимальной электрофоретической подвижностью, и концевой электролит, содержащий ион с минимальной подвижносгью ионов анализируемой смеси А, В, С и D). Под воздействием электрического поля смесь разделяется на примыкающие друг к другу зоны индивидуальных компонентов, выходящих в порядке понижения их электрофоретической подвижности: лидирующий ион, А, В, С, D и концевой ион. Возможность регулирования концентраций в исследуемых зонах за счет изменения свойств лидирующего электролита и равномерность концентрации данного компонента внутри каждой зоны делаег этот метод особенно ценным для качественного и количественного анализа и препаративного разделения. В гель-электрофорезе смесь заряженных макромолекул разделяется в результате различия в их заряде, размере и скорости миграции через гель, помещенный в электрическое поле. При этом на электрофоретическую миграцию молекул оказывает влияние матрица геля и, поэтому достигается селективное разделение молекул по размерам. Гели на основе крахмала, агарозы или силикагеля используют в первую очередь для стабилизации зон. Макропористые гели, особенно гели агара и агарозы, наиболее широко применяют в методе иммуномектрофореза . Особенностью этого метода является обработка смеси белков раствором антисыворотки после проведения электрофоретического разделения. Антисыворотка содержит в себе специфические антитела, воздействующие на индивидуальные компоненты смеси белков. Антигены и антитела, взаимодействуя между собой, оседают, образуя линии осаждения, каждая из которых должна соответствовать определенному белку. Иммуноэлектрофорез позволяет проводить идентификацию белков серологическим и физико-химическими методами. Путем введения радиоактивной метки в антигены можно существенно повысить чувствительность метода Эта разновидность электрофореза получила название радиоиммуноэлектрофорез. Полиакриламидный гель химически весьма инертен. Его слабое сродство к красителям позволяет быстро обнаруживать биополимеры (белки, нуклеиновые кислоты) по цветным реакциям. Характерен мегод электрофореза в градиен 1.4.15. Капиллярный электрофорез Одним из самых современных и перспективных сепарационных методов на сегодняшний день является капиллярный электрофорез, вобравший в себя все лучшие качества хроматографических методов и электрофореза. Развитие этого метода началось в конце 70-х, начале 80-х годов прошлого века с работ Миккерса, Эвериртса, Поргенсона и Лукаса. В этом методе разделение смеси заряженных или нейтральных молекул основано на различии в их электрофоретичеекой подвижности и (или) распределении между раствором и движущимися в электрическом поле заряженными частицами (мицеллами, коллоидными частицами, металлокомплексами, молекулами полиэлектролита). При анализе методом капиллярного электрофореза пробу небольшого объема (несколько нанолитров) вводят на анодном конце в кварцевый капилляр, заполненный ведущим электролитом (например, фосфатные и боратные буферные растворы с концентрациями 10-100 мМ, растворы органических и неорганических солей, кислот и оснований). К тонкому кварцевому капилляру (внутренний диаметр 25-100 мкм) длиной 20-100 см прикладывают напряжение от 10 до 30 кВ. Под действием электрического ноля компоненты пробы начинают двигаться по капилляру с разной скоростью, зависящей or их структуры, заряда и молекулярной массы, и соответственно, в разное время достигают детектора. Разделение пробы достигается приложением напряжения к буферным сосудам, на электрофоретическое перемещение всегда накладывается более или менее интенсивный электроосмотический поток, способствующий пассивной транспортировке зоны пробы, а не ее разделению. Этот эффект сильно зависит от рН буфера и от свойств поверхности капилляра, Он может быть настолько большим, что будут двигаться не только нейтральные молекулы, но даже отрицательно заряжениые ионы, которые могут перемешаться к детектору, несмотря на их злектфофоретическую миграцию. На поверхности кварцевого капилляра из-за диссоциации силанольных трупп образуются в процессе электрофореза отрицательные заряды, вблизи стенок индуцируются положительные заряды, и электроосмотический поток будет направлен к катоду. Это обуславливает расположение детектора вблизи катодного пространства. Благодаря электроосмотическому потоку к катоду все незаряженные молекулы перемещаются с одинаковой скоростью и не могут быть разделены, в то время как разделение заряженных ионов возможно благодаря их различной электрофоретической подвижности. Элекгроосмотический поток можно регулировать с помощью химического модифицирования поверхности капилляра, при этом одновременно уменьшается возможная адсорбция компонентов пробы на поверхности капилляра и улучшается воспроизводимость анализа. В качестве покрытий используются алкилсилановые, фторалкильные, гидрофильные полиэфирные, полиакриламидные и другие покрытия. Детектирование компонентов пробы может осуществляться спек-трофотометрическим, кондуктометрическим, рефрактометрическим, спектрофлюорометрическим или масс-сиектромефическим детектором. Детектор расположен ближе к катодному концу капилляра. Капилляр принудительно охлаждается. Способ ввода пробы гидродинамический (созданием давления в сосуде для пробы или созданием вакуума в катодном буферном резервуаре) или за счет электрофоретической миграции пробы в капилляр в результате приложения соответствующего электрического напряжения к концам капилляра. Так, в системе капиллярного электрофореза " Капель-103" ввод пробы осуществляют гидродинамически при 900 мбар'с. На рис. 1, 29. представлена схема аппарата для капиллярного электрофореза. Полученная электрофореграмма представляет собой последовательность пиков, по которым, как и в хроматограмме, можно идентифицировать и количественно определить конкретное соединение. Boспроизводимость времен удерживания - 5%. Среднее квадратическос отклонение выходного сигнала по высоте пика - 5 %. Пределы обнаружения веществ 0.2 - 1.0 мг/дм3. Обработка результатов проводится с помощью типовых хроматографических программ.
Pис. 1.29. Принципиальная схема капиллярного электрофоризера: 1 - анодная зона, вход капилляра; 2 - катодная зона, выход капилляра; 3 -детектор; 4 - источник высокого напряжения; 5 -регистрирующее устройство; 6 - контейнеры для проб, 7, 8 - сосуды с буферным раствором
Популярное: |
Последнее изменение этой страницы: 2016-03-16; Просмотров: 1635; Нарушение авторского права страницы



 (1.37)
(1.37) (1.38)
(1.38)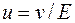 (1.39)
(1.39)
 те плотности полиакриламидного геля. На первом этапе осуществляют зонный электрофорез, в заключение - молекулярно-ситовое разделение. Движение веществ от старга постепенно тормозится вследствие уменьшения пор геля. Ограничение подвижности, вызываемое гелем, приводит к фокусированию зон.
те плотности полиакриламидного геля. На первом этапе осуществляют зонный электрофорез, в заключение - молекулярно-ситовое разделение. Движение веществ от старга постепенно тормозится вследствие уменьшения пор геля. Ограничение подвижности, вызываемое гелем, приводит к фокусированию зон.
