
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Физическая химия неравновесных процессовСтр 1 из 12Следующая ⇒
Физическая химия неравновесных процессов (курс лекций)
Екатеринбург Содержание
Введение 4 I. Химическая кинетика 5 Основные понятия химической кинетики 5 1.1. Классификация химических реакций 5 1.2. Элементарные химические реакции 6 1.3. Скорость химических реакций 6 1.4. Кинетические кривые и уравнения 7 1.5. Молекулярность и порядок реакции 8 Элементарный акт химического превращения 9 2.1. Теория переходного состояния или активированного комплекса 9 2.2. Односторонние (необратимые) реакции первого порядка 11 2.3. Односторонние реакции второго порядка 13 2.4. Односторонние реакции n-го порядка 14 2.5. Методы определения порядка реакции 15 Сложные химические реакции 17 3.1. Обратимые (двусторонние) химические реакции 18 3.2. Параллельные химические реакции 19 3.3. Односторонние последовательные реакции 21 Зависимость скорости химической реакции от Температуры 23 Гетерогенные химические реакции 25 Каталитические реакции (катализ) 29 6.1. Свойства катализаторов 29 6.2. Каталитическая активность и избирательность 32 6.3. Кислотно-основной катализ 33 6.4. Гетерогенный катализ. Теории гетерогенного катализа 36 II. Электрохимия 41 Общие понятие об электрохимических системах 41 1.1. Предмет электрохимии 41 1.2. Проводники электрических зарядов 41 1.3. Электролиз. Законы Фарадея. 41 Растворы электролитов, их свойства 43 2.1. Сильные и слабые электролиты 43 2.2. Электролитическая диссоциация в растворе 44 2.3. Механизм переноса тока в растворах электролитов. Числа переноса 47 2.4. Удельная и эквивалентная электрические проводимости растворов электролитов 48 2.5. Молярная электрическая проводимость 50 2.6. Молярная электрическая проводимость ионов гидроксония и гидроксида 51 2.7. Влияние природы растворителя на предельную подвижность ионов 53 2.8. Зависимость молярной электропроводимости от температуры 54 Ионные равновесия в растворах электролитов 56 3.1. Термодинамическая константа диссоциации растворов электролитов 56 3.2. Диссоциация воды. Ионное произведение воды 59 Электродные процессы и электродвижущие силы 61 4.1. ЭДС электрохимической системы, электродный потенциал 61 4.2. Строение двойного электрического слоя на границе раствор – металл 62 4.3. Устройство и изображение гальванического элемента 63 4.4. Компенсационный метод измерения ЭДС 65 4.5. Термодинамика гальванического элемента 65 4.6. Вывод уравнения Нернста для расчёта ЭДС гальванического элемента 67 Введение
Основными задачами учебной дисциплины «Физическая химия неравновесных процессов» является изучение и объяснение закономерностей, определяющих временные характеристики химических процессов, скорость их протекания, влияние на них параметров окружающей среды, условия получения максимального выхода продукта. Эта дисциплин формирует творческое инженерное мышление студентов, стремится к количественному описанию химических процессов, используя такие фундаментальные методы исследования как термодинамический, квантово – химический, статистический, поэтому для успешного усвоения кинетических закономерностей химических превращений студент должен владеть основами физики и высшей математики, а также иметь твердые знания по неорганической и органической химии. Знание физико – химических закономерностей протекания химических реакций во времени открывает перед инженером широкие возможности для понимания функционирования экологических систем, разработки новых технологических процессов, удовлетворяющих современным требованиям экологической безопасности, для разработки методов и средств, необходимых для создания автоматизированных систем мониторинга окружающей среды. Преподавание физической химии неравновесных процессов имеет цель дать студенту тот фундамент, с помощью которого можно провести количественный анализ любого физико-химического процесса. I. Химическая кинетика
Основные понятия химической кинетики Химическая кинетика – это наука, изучающая протекание химической реакции или физико – химических процессов во времени, это раздел физической химии, в котором изучается зависимость скорости химической реакции от концентрации реагентов, температуры, свойств среды, излучения и других факторов. Химическая кинетика позволяет рассчитать время заданной степени превращения вещества и найти наиболее эффективные факторы воздействия на скорость химической реакции.
Элементарный акт химического превращения Сложные химические реакции
Под сложными реакциями понимают реакции, протекающие в несколько стадий, 2х – сторонние, параллельные, последовательные. При описании сложных реакции учитывается условие материального баланса и принцип независимого протекания реакции; каждая реакция протекает независимо от другой и скорость каждой реакции пропорциональна концентрации реагирующих веществ. Условно сложную реакцию разбивают на несколько элементарных стадий. Зависимость скорости химической реакции от температуры. Уравнение Аррениуса. Скорость химической реакции зависит не только от концентрации реагентов, но и от температуры. По правилу Вант-Гоффа увеличение температуры на 10о приводит к увеличению скорости реакции в 2-4 раза.
Более строгая зависимость k от T выражается уравнением Аррениуса:
где A и B – const; A – предэкспоненциальный множитель, B=
Другая запись уравнения Аррениуса:
Для газофазных реакций A = 1012 -1016C-1
Строят график в координатах
Свойства катализаторов
Свойства катализаторов: (kt) 1) катализатор не влияет на положение термодинамического равновесия, т.е на величину константы равновесия
2) катализаторы обладают избирательностью (или селективностью действия). Каждый катализатор может ускорять лишь некоторые реакции. Ni – катализатор реакции гидрирования V2O5 – катализатор реакции окисления Для каталитического действия обычно достаточно малое количество катализатора. Влияние kt на скорость процесса характеризуется удельной каталитической активностью, за меру которой принимается скорость реакции в присутствии kt, отнесенная к единице количества kt (гомогенный катализ) или к единице поверхности kt (гетерогенный катализ). Механизм действия kt очень сложный. В присутствии kt: 1) снижается величина Eакт; 2) увеличивается скорость реакции; 3) kt может вызвать цепные реакции; 4) делает более вероятным протекание некоторых реакций. Рассмотрим влияние катализатора на кинетику химических реакций на примере гомогенно-каталитической бимолекулярной односторонней реакции
Константа равновесия образования активного комплекса
Скорость реакции в присутствии катализатора
Из уравнения (1.86) значение
Концентрация свободного катализатора
Из уравнения (1.86) выразим отношение
Подставим это значение
Уравнение (1.90) показывает, что скорость гомогенно-каталитической бимолекулярной реакции пропорциональна концентрации катализатора Анализ уравнения: 1) при малых значениях
Общий порядок реакции по исходным веществам – второй, а по веществам A или B – первый. 2) если значение
При этом реакция протекает по нулевому порядку (по исходным веществам). 3) при средних значениях Если каталитическая реакция
протекает в две стадии с образованием неустойчивого промежуточного соединения AK
то скорость реакции выражается уравнением (1.90)
В присутствии катализатора может протекать термодинамически возможная реакция по новому пути с образованием других продуктов
или
При образовании активного комплекса в присутствии катализатора энтропия активации уменьшается в присутствии kt по сравнению с энтропией активации без kt. Это приводит к уменьшению константы скорости реакции. Т.о., увеличение константы скорости реакции может быть связана только с уменьшением энергии активации реакции. Константа скорости реакции образования активного комплекса может быть рассчитана по формуле
k - const Больцмана h - const Планка T - температура
Кислотно-основной катализ Для неводных растворов неприменимы обычные представления о кислотах и щелочах. Согласно обобщенной теории протолитического равновесия Бренстеда: кислота – химическое соединение, способное в течении реакции отдавать протон – донор протонов; Реакция присоединения протона – протонизация. Соединения
Основание – химическое соединение, способное присоединять протон – акцептор протона Отнятие протона – депротонизация
В отщепление или присоединении протона участвует пара сопряженных кислоты и основания.
Одно и тоже вещество может выступать в роли кислоты и основания. Например, анилин в воде отнимает протон от молекулы воды и выступает как основание.
В жидком аммиаке (как растворителе) анилин является кислотой, отдает протон
Кислотно-основной катализ - наиболее распространенный и практически важный вид гомогенного катализа. При кислотно-основном катализе две основных стадии: при кислотном катализе: 1. протолитическая реакция и протонизация исходного вещества S за счет передачи протона от kt – кислоты AH; 2. катализатор – кислота регенерируется после образования продуктов реакции P и Q для мономолекулярной реакции
В кислотном катализе kt может быть любое вещество, способное отщеплять протон (ион при основном катализе: 1 стадия – протолитическая реакция между исходным веществом SH и основанием B – депротонизация исходного вещества или передача протона kt-основанию
2 стадия – депротонизированная форма исходного вещества распадается с образованием продуктов P и Q и регенерацией kt kt – любое вещество, присоединяющее протон; например: ион Лимитирующей стадией кислотно-основного катализа может быть первая или вторая стадия. Рассмотрим кислотный катализ, когда первая стадия – протонизация близка к равновесному процессу, вторая – лимитирующая:
Вторая стадия протекает по кинетическому уравнению реакции I порядка. Скорость реакции определяется по скорости II лимитирующей стадии:
где k – истинная константа скорости кислотно-каталитического превращения Равновесная концентрация протонной формы вещества
Тогда
Зависимость k от Выразим долю α : Для I равновесной стадии
Учитывая, что
После небольших преобразований получим
Величину В разбавленных растворах
В концентрированных растворах кислот значения Общая концентрация вещества в растворе
Вычислим
Анализ уравнения:
1) если то
Зависимость
2) если Опытное значение 3) если
При кислотно-основном катализе, когда I стадия считается лимитирующей (протонизация), согласно дуалистической теории кислотно-основного катализа Даусона при расчете скорости каталитического процесса необходимо учитывать, что каталитически активными центрами являются ионы гидроксония и гидроксила, а также молекулы недиссоциируемых кислот и оснований, недиссоциируемые молекулы воды. Скорость реакции равна сумме всех скоростей реакции, обусловленных всеми катализирующими частицами:
Где
При добавлении к раствору соли MA, скорость реакции возрастает за счет слагаемого II Электрохимия Предмет электрохимии
Электрохимия – это раздел физической химии, в котором изучаются законы взаимного превращения химической и электрической формы энергии и систем, где эти превращения совершаются. Электрические системы и явления рассматриваются в равновесных условиях в отсутствии электрического тока и в неравновесных при прохождении электрического тока. В разделе электрохимии изучаются физико-химические свойства ионных проводников, процессы и явления на границе раздела фаз с участием заряженных частиц – ионов и электронов. Электрохимия, как наука, возникла на рубеже 18-19 веков на основании трудов Л. Гальвани, А. Вольта, В. Петрова, Г. Деви и М. Фарадея. Электрохимия имеет большое практическое значение, а именно: электролиз в металлургии легких и цветных металлов, химическая промышленность, технология гальванотехники, коррозия и защита металлов.
Электролиз. Законы Фарадея.
Взаимное превращение электрической м химической энергии происходит в электрохимических системах, состоящих из следующих частей: 1. Проводники II рода; 2. Проводники I рода – металлические электроды. Положительный электрод называется анодом, отрицательный – катодом. 3. Проводники I рода – внешняя электрическая цепь, связывающая электроды. Электролиз – это химическое превращение в электрической системе под действием внешнего электрического поля. При электролизе отрицательные ионы движутся к аноду (+) – анионы. Положительно заряженные частицы – катионы – двигаются к катоду. Вещества, которые в растворе (или расплаве) состоят полностью или частично из ионов называются электролитами. Представим систему, в которой имеется раствор медного купороса (CuSO4 + H2O). При растворении в воде электролита происходит диссоциация на ионы CuSO4 ⇆ Сu2+ + SO42- При погружении в раствор электродов (катода и анода) начинается направленное движение ионов. На катоде происходит восстановление меди по реакции: Cu2+ + 2e → Cu0 и выделяется на нем металлическая медь. Соотношение между (количеством) массой прореагировавших веществ и количеством постоянного электрического выражается законами Фарадея. I закон Фарадея: Масса вещества m претерпевшего химическое превращение под действием электрического тока пропорциональна количеству протекшего электрического тока.
m – масса вещества; J – сила тока; τ – время электролиза Kэ – электрохимический эквивалент.
II закон Фарадея: При прохождении через различные электролиты одного и того же количества электричества массы различных веществ, участвующих в реакциях, пропорциональны их химическим эквивалентам. Для выделения 1 г –экв. любого вещества требуется пропустить 1 Фарадей электричества.
1 Фарадей = 1F = 96500 Кл
Э – химический эквивалент, M – мольная масса вещества; z – число
тогда II закон Фарадея дает физический смысл Kэ: Кэ – это количество вещества, которое претерпевает превращение при пропускании через раствор 1 Кл электричества. На практике часто наблюдается отклонение от законов Фарадея вследствие протекания побочных реакций; а именно: 1). в электролитах, содержащих кислоты, происходит дополнительно восстановление водорода (кислота увеличивает электропроводность) Например: электролит ZnSO4 + H2SO4 на катоде(-): Zn2+ + 2e → Zn0 – основная 2Н+ + 2е→ H2 ↑ – побочная 2). Неполное восстановление металла на катоде: Fe3+ + 3e→ Fe0 Fe3+ + 1e→ Fe2+ 3). Наличие нескольких реакций восстановления на катоде: Au3+ + 3e→ Au0 – основная Au1+ + 1e→ Au0 - побочная 4). Взаимодействие продуктов электролиза с электролитом: H2O + NaCl H2O ⇆ H+ + OH- Ионы щелочных металлов не восстанавливаются, на катоде идет восстановление водорода: (-) 2H2+ + 2e = H2↑ В прикатодном пространстве накапливаются ионы натрия, в прианодном - ионы гидроксила На аноде идет окисление CI— ионов: (+) 2CI− - 2e → CI2↑ выделяется газ. Образующаяся NaOH взаимодействует с CI2: 2NaOH + CI2 = NaCI + NaCIO + H2O Эффективность электрохимического процесса оценивается величиной выхода по току
В отсутствии побочных процессов ψ =1, но на самом деле ψ < 1.
Числа переноса
В растворе электролита сольватированные ионы находятся в беспорядочном тепловом движении. При наложении электрического поля возникает упорядоченное движение ионов к противоположно заряженным электродам – миграция (перенос).
Е – приложенное напряжение постоянного тока, В, h – расстояние между электродами, Vi – скорость иона, Чем выше приложенное напряжение Е, тем быстрее двигается ион; чем больше расстояние между электродами, тем меньше скорость движения иона. Под скоростью движения иона Vi понимают расстояние, которое проходит ион в единицу времени в направлении одного из электродов.
Абсолютная скорость движения (подвижность) ионов ui не зависит от Е и h и определяется соотношением
Абсолютная скорость иона определяется природой иона, Т и не зависит от условий проведения процесса
Произведение
Каждый вид иона переносит определенное количество электричества. Для оценки доли участия данного вида иона в переносе электричества Гитторфом введено понятие о числе переноса. Число переноса иона i – вида – это отношение количества электричества, перенесенного ионом данного вида, к общему количеству электричества
Можно найти значение чисел переноса иона как отношение
Сумма чисел переноса всех видов ионов в растворе равна 1.
Поэтому для расчетов достаточно задать одно значение числа переноса и рассчитать другое значение.
Гидроксония и гидроксида
В водных растворах протон (ион) водорода записывают в виде иона гидроксония – H3O+ H+ + H2O ⇆ H3O+ Ионы H3O+ и гидроксида OH− обладают более высокой молярной электрической проводимостью или подвижностью, чем другие ионы. Подвижности большинства катионов и анионов лежат в пределах (40-80) 10-4 Значение предельной подвижности иона гидроксония 1. При наложении электрического поля переход протона от H3O+ к молекуле воды повышается в направлении поля. Передвижение протона совершается по цепочке от одной молекулы воды к другой по схеме:
Электричество переносится мигрирующими ионами H3O+ и протонами. 2. Аналогичным образом объясняется подвижность ионов гидроксида. Протон переходит от молекулы воды к иону гидроксида
Так как энергия отрыва протона от молекулы воды больше, чем от иона гидроксония, то вероятность перехода протона от H2O к OH− меньше, чем от Н3O+ к воде. Поэтому Предельные подвижности некоторых ионов Р = 1 атм, t = 25°С
При одном и том же заряде иона с увеличением радиуса иона увеличивается подвижность иона, так как на поверхности иона уменьшается плотность заряда, уменьшается степень его гидратации и эффективный радиус иона.
Подвижность ионов
Толь в водных растворах и спиртах сохраняется высокая подвижность ионов H+ (протонов), а OH- в воде. Зависимость молярной электропроводности ионов от вида растворителя.
При взаимодействии HCI со спиртом образуется катион этоксоний (общее название меоксоний)
этоксоний
HCI + H2O = H3O+ + CI- гидроксоний
Эстафетный механизм передачи протона вдоль цепочки молекул растворителя сохраняется и в спиртах. Подвижность ионов в различных растворителях зависит от диэлектрической проницаемости растворителя. Чем выше диэлектрическая проницаемость (ε ) растворителя, тем выше степень диссоциации электролита и электрическая проводимость раствора.
Молярная электрическая проводимость HCI в различных растворителях (t=250C, p=1атм.)
Растворов электролитов
Константа равновесия любого химического процесса, в том числе и диссоциации, выраженная через активность, записывается следующим образом:
Из уравнения изотермы химической реакции активность иона Для примера рассмотрим диссоциацию одноосновной кислоты в водном растворе: HA + H2O ⇆ H3O+ + A- Протон Н+ в природе не существует, в водных растворах он всегда гидратирован молекулами и существует в виде иона гидроксония.
Обычно в растворах С0, 1≫ С0, 2, т.е. концентрация растворителя значительно больше концентрации растворенного вещества.
Кдисс включает в себя произведение Кравн. на активность воды.
Перегруппируем
Активности катиона и аниона определить невозможно; активность молекулярной формы принято считать равной единице. Тогда
Известно, что
Для одноосновной кислоты
С учетом этого
Кс – концентрационная константа диссоциации, которая зависит от С, Т и природы растворителя. Выразим Кс через степень электролитической диссоциации α : НА +Н2О ⇆ Н3О+ +А⁻
Степень диссоциации электролита
Отсюда
Значение константы Кс можно определить экспериментальным путем по значению α :
λ ∞ рассчитывают по уравнению Кольрауша:
Популярное:
|
Последнее изменение этой страницы: 2017-03-11; Просмотров: 811; Нарушение авторского права страницы
 - температурный коэффициент скорости химической реакции
- температурный коэффициент скорости химической реакции (1.64)
(1.64) ;
;  - энергия активации химической реакции
- энергия активации химической реакции
 (1.65)
(1.65)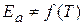 , а зависит от природы реакции. Пользуясь этим уравнением можно определить энергию активации, если известны k при различных температурах.
, а зависит от природы реакции. Пользуясь этим уравнением можно определить энергию активации, если известны k при различных температурах. - величина потенциального барьера.
- величина потенциального барьера.
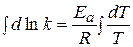
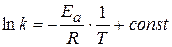
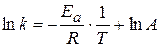 (1.66)
(1.66)

 (1.67)
(1.67) < 0;
< 0; 


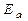 =
= 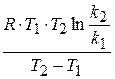 (1.68)
(1.68) (1.85)
(1.85)

 равна
равна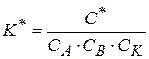 (1.86)
(1.86) - концентрация активных комплексов;
- концентрация активных комплексов;  - концентрация исходных веществ;
- концентрация исходных веществ;  - концентрация катализатора, не вошедшего в активный комплекс.
- концентрация катализатора, не вошедшего в активный комплекс. (1.87)
(1.87)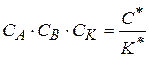 , его подставим в уравнение (1.87), тогда
, его подставим в уравнение (1.87), тогда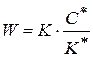 (1.88)
(1.88)
 - общая или исходная концентрация катализатора
- общая или исходная концентрация катализатора :
: 
 (1.89)
(1.89) в уравнение скорости реакции
в уравнение скорости реакции (1.90)
(1.90) можно принять, что сумма
можно принять, что сумма  , тогда уравнение (1.90) можно записать
, тогда уравнение (1.90) можно записать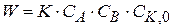 (1.91)
(1.91) и скорость реакции
и скорость реакции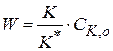 (1.92)
(1.92)

 (1.93)
(1.93) и
и  , которые без катализатора практически не образуются
, которые без катализатора практически не образуются

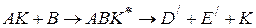
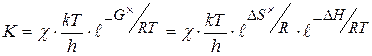 (1.94)
(1.94) - изобарный потенциал активации
- изобарный потенциал активации - трансмиссионный коэффициент прохождения (≈ 1), для приближенных расчетов можно не учитывать
- трансмиссионный коэффициент прохождения (≈ 1), для приближенных расчетов можно не учитывать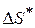 - энтропия образования активного комплекса
- энтропия образования активного комплекса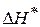 - энтальпия образования активного комплекса
- энтальпия образования активного комплекса - зависит от энтальпии и энтропии активации.
- зависит от энтальпии и энтропии активации.



 и
и  - сопряженные кислота и основание.
- сопряженные кислота и основание.


 - соль
- соль  )
)
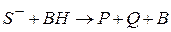
 (соль
(соль  )
) (I)
(I)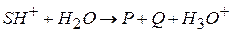 (II) – лимитирующая
(II) – лимитирующая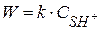
 составляет долю от общей концентрации
составляет долю от общей концентрации  вещества S в растворе:
вещества S в растворе: 
 (1.98)
(1.98)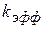 - эффективная константа скорости катализа
- эффективная константа скорости катализа ;
; 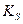 - константа кислотности вещества S
- константа кислотности вещества S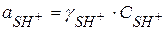

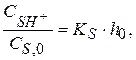 где
где 
 называют кислотностью среды, она характеризует способность отдавать протон.
называют кислотностью среды, она характеризует способность отдавать протон. и
и  отсюда
отсюда и
и  ;
;  - функция кислотности Гамета
- функция кислотности Гамета и
и  отличаются на несколько порядков, поэтому в концентрированных растворах нужно пользоваться понятиями кислотности среды и функции кислотности
отличаются на несколько порядков, поэтому в концентрированных растворах нужно пользоваться понятиями кислотности среды и функции кислотности  и
и 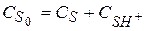
 и
и 
 (1.99)
(1.99)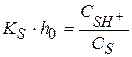
 и
и 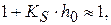

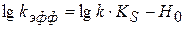
 от
от  . По известному значению
. По известному значению  находят k.
находят k. с
с 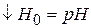
 и
и 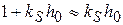 , то
, то 
 не должно зависеть от константы кислотности
не должно зависеть от константы кислотности 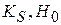 и
и 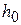
 , можно уравнение записать в следующем виде
, можно уравнение записать в следующем виде (1.100)
(1.100)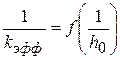 носит линейный характер, графически определяем k и KS
носит линейный характер, графически определяем k и KS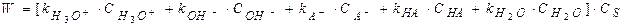
 - константы скорости параллельных реакций;
- константы скорости параллельных реакций;  - концентрация катализаторов.
- концентрация катализаторов. . Этот эффект называется вторичным солевым эффектом.
. Этот эффект называется вторичным солевым эффектом. , где
, где  (2.1)
(2.1) – количество электричества.
– количество электричества.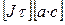 =[кулон]
=[кулон] (2.2)
(2.2)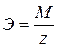 ;
;  , принимающих участие в электродной реакции.
, принимающих участие в электродной реакции. , (2.3)
, (2.3) – электрохимический эквивалент
– электрохимический эквивалент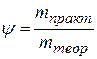 (2.4)
(2.4)
 .
.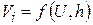 (2.12)
(2.12) - градиент потенциала или напряженность.
- градиент потенциала или напряженность.
 на число Фарадея называется подвижностью иона – λ i.
на число Фарадея называется подвижностью иона – λ i.

 (2.13)
(2.13) (2.14)
(2.14) (2.15)
(2.15) (2.16)
(2.16) . Значение предельной подвижности, например, иона натрия
. Значение предельной подвижности, например, иона натрия 







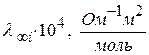


 HCI + C2H5OH = C2H5OH2+ +CI-
HCI + C2H5OH = C2H5OH2+ +CI-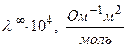
 (2.38)
(2.38) .
.  (Т, природы растворителя).
(Т, природы растворителя). (2.39)
(2.39)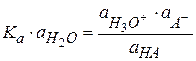 (2.40)
(2.40)
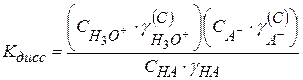 (2.41)
(2.41) (2.42)
(2.42) (2.43)
(2.43)

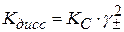 (2.44)
(2.44)

 - это уравнение является законом разведения Оствальда.
- это уравнение является законом разведения Оствальда.
