|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Сопротивления стадий ионизации и диффузии.
Сопротивления, или поляризуемости, анодного и катодного процессов Ра и Рк складываются из сопротивления реакций Рр и сопротивления диффузии Рд, т.е.
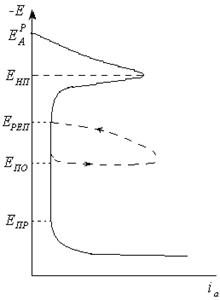
Рис. 4.18. Поляризационная коррозионная диаграмма.
предельного диффузионного тока (на примере катодного процесса кислородной деполяризации) Iд с помощью следующих уравнений:
Тогда Если катодный процесс состоит из двух параллельно идущих катодных реакций - ионизации кислорода и выделения водорода (рис.4.18) (смешанная кислородно-ионизации кислорода и выделения водорода (смешанная кислородно-водородная деполяризация), то анодная и катодная поляризационные кривые пересекутся в точке К и степень контроля катодного процесса в этом случае характеризуется соотношением силы коррозионного тока, определяемого процессом ионизации кислорода
где I2 - коррозионный ток. Значения Практические случаи контроля коррозионных процессов На практике встречаются шесть основных случаев контроля электрохимических коррозионных процессов, приведенных на рис. 4.19: а) катодный контроль при основной роли перенапряжения ионизации кислорода (R≈ 0, ∆ Ек> > ∆ Еa, Рр> Pд). Наблюдается при коррозии металлов в электролитах при хорошем подводе кислорода к поверхности корродирующего металла; б) катодный контроль при основной роли диффузии кислорода (R≈ 0, ∆ Ек> > ∆ Еa, Рд> Pр). Наблюдается при коррозии железа, цинка и других металлов в неперемешиваемых нейтральных электролитах; в) катодный контроль при основной роли перенапряжения водорода (R≈ 0, ∆ Ек> > ∆ Еa,
Рис. 4.19. Поляризационные коррозионные диаграммы для основных практических случаев контроля электрохимических коррозионных процессов.
г) смешанный анодно-катодный контроль (R≈ 0, ∆ Еа≈ ∆ Ек). Наблюдается при коррозии железа, нержавеющих сталей, алюминия и других металлов в пассивном состоянии; д) смешанный катодно-омический контроль (R≠ 0, ∆ Ек≈ ∆ ЕR> > ∆ Еа). Наблюдается при коррозии вследствие работы макропар на больших расстояниях в электролитах с очень низкой электропроводностью (при коррозии подземного трубопровода вследствие работы макропар неравномерной аэрации); е) смешанный катодно-анодно-омический контроль (∆ Ек≈ ∆ ЕR≈ ∆ Еа). Наблюдается при легком доступе кислорода к поверхности корродирующего металла, склонности к пассивированию и при большом сопротивлении электролита (при влажной атмосферной коррозии сталей под очень тонкой пленкой влаги). Определение контролирующего процесса электрохимической коррозии металла важно, так как для уменьшения скорости коррозии наиболее эффективным обычно является воздействие на контролирующий процесс с целью затруднить его протекание. Пассивность металлов Явление пассивности, впервые открытое М.В. Ломоносовым, имеет очень большое практическое значение в металлургии, и, особенно, для защиты металлов от коррозии. На его принципах существует целая отрасль металлургии – коррозионностойкие стали (хромистые, хромоникелевые, хромомолибденовые, и т.п.) и сплавы (алюминиевые, никелевые, титановые и т.д.). Пассивность (пассивное состояние) – состояние относительно высокой коррозионной стойкости, вызванное торможением анодной реакции ионизации металла в определенной области потенциала. Скорость коррозии сталей и сплавов при переходе в пассивное состояние может понижаться на несколько порядков (рис. 4.20).
Рис. 4.20. Зависимость логарифма скорости коррозии железа от концентрации HNO3 при 200С.
М. Фарадеем было доказано, что явление пассивности имеет электрохимическую природу. Он разработал пленочную теорию пассивности, согласно которой в ходе анодной реакции образуется тонкая невидимая оксидная плёнка, сильно замедляющая анодный процесс. Согласно этой теории пассивные плёнки - тонкие беспористые плёнки оксидов с относительно высокой электронной, но низкой ионной проводимостью. Последнее условие обязательно, т.к. на железе в электролитах при определённых условиях могут образовываться слои толстых рыхлых кислородсодержащих соединений, не обладающих защитными свойствами. Плёночная теория применима ко многим металлам (Al, Ni, Cr, Ti, редкие тугоплавкие металлы). Образование пассивных пленок обусловлено анодной реакцией образования оксида в присутствии H2O:
Адсорбционная теория пассивности (А.Н. Фрумкин) состоит в том, что поверхностные атомы металла связываются с молекулами H2O адсорбционной связью, прочность которой увеличивается с ростом потенциала. Плёночно - адсорбционная теория, объединяя предыдущие, дополняет их возможностью нахождения под плёнкой и в порах хемосорбированных ионов кислорода. Основной метод изучения пассивности металла – потенциостатический, т.е. получение электрохимической зависимости скорости растворения металла от потенциала с использованием потенциостата, когда металлу задается потенциал и фиксируется ток. Типичная анодная поляризационная кривая приведена на рис. 4.21. Другим методом получения поляризационных зависимостей является гальваностатической метод, при котором задается ток, а регистрируется изменение потенциала.
а б Рис.4.21. Обобщённая анодная потенциостатическая поляризационная кривая при пассивации металла (а). Характерные потенциалы:
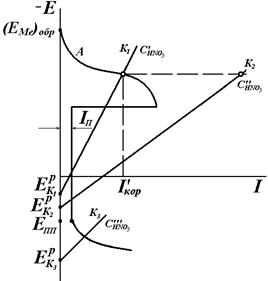
На рис. 4.21, а приведена обобщенная анодная поляризационная Е – i кривая. Здесь Условие (4.90) не является достаточным для пассивации конкретного металла. Например, в растворах HNO3 переход железа в пассивное состояние зависит от концентрации HNO3 (рис. 4.21, б). Катодная кривая К1 (восстановление HNO3) пересекает анодную кривую А в области активного растворения при низкой концентрации
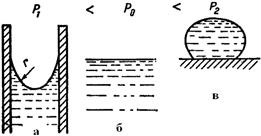
Для более высокой концентрации, Пассивация металла может быть достигнута: 1) созданием условий для самопроизвольной пассивации в присутствии окислителей, 2) анодной поляризацией металла до потенциалов пассивной области. При изменении внешних условий пассивный металл может перейти в активное состояние. Факторами активации (депассивации) металла являются: 1) присутствие восстановителей (водорода, солей Na2SO3, Na2S2O8 и др.); 2) катодная поляризация от внешнего источника постоянного тока или контакт пассивного металла с металлами, являющимися анодом; 3) влияние некоторых ионов (H+, или Cl- на пассивный металл); 4) повышение температуры; 5) механическое нарушение пассивной поверхности металла, например, царапание, если металл не находится в пассивирующей среде. Атмосферная коррозия Атмосферной коррозией называют коррозионное разрушение металлов под воздействием окружающей воздушной среды. Это наиболее распространенный вид коррозии, которому подвергаются в процессе эксплуатации машины, оборудование и сооружения при контакте с влажной и загрязненной атмосферой. Атмосферная коррозия - коррозия металла в атмосфере воздуха (электрохимическая коррозия, протекающая в условиях любого влажного газа). Основным фактором, определяющим механизм и скорость атмосферной коррозии, является влажность поверхности металлоконструкций. Образование пленки влаги зависит от относительной влажности воздуха, температуры поверхности металла, атмосферных осадков (при эксплуатации на открытом воздухе), наличия в атмосфере гигроскопических продуктов, состояния поверхности и пористости материала (рис.4.22.1).
1 2 Рис. 4.22. 1 - зависимость скорости атмосферной коррозии от толщины слоя влаги на поверхности металла: I – сухая, II – влажная, III - мокрая, IV – при полном погружении, 2 – влияние формы поверхности жидкости на давление насыщенных паров (а – вогнутый, б – плоский, в – выпуклый мениски. По степени увлажненности различают атмосферную коррозию: - мокрую – при наличии на поверхности видимой пленки влаги (относительная влажность воздуха ~100 %, непосредственное увлажнение поверхности дождем, туманои, сопровождающемся образованием видимых фазовых слоев воды; - влажную – при относительной влажности воздуха 60-70%, когда становится возможной конденсация влаги, проявляющаяся в появлении на поверхности адсорбционной пленки воды; - сухую – коррозия при полном отсутствии пленки влаги на поверхности металла; реализуется при относительной влажности воздуха мене 60 %, когда на поверхности отсутствуют следы влаги. Поскольку в условиях эксплуатации возможно изменение внешних условий, вызывающих переход одного типа атмосферной коррозии в другой, деление достаточно условно. Вместе с тем, три типа состояний значительно отличаются по механизму коррозионного процесса. При сухой коррозии реализуется химический механизм процесса: на поверхности металла формируются тонкие защитные оксидные пленки, тормозящие развитие коррозии. Рост пленок происходит в начальный момент контакта со средой, в последующем роста толщины пленки практически не происходит. Максимальная толщина пленок на железе – 3-4 нм, на нержавеющих сталях – 1-2 нм. При влажной коррозии, с образованием адсорбированного слоя влаги на поверхности, реализуется электрохимический механизм. Скорость коррозии значительно возрастает. При небольшой толщине этого слоя (10 - 1000 нм) кислород проникает через него к поверхности металла, что обеспечивает протекание с высокой скоростью катодного процесса, который протекает с анодным контролем. При образовании фазовых пленок в области мокрой коррозии затруднятся доступ кислорода к катодным участкам поверхности, что вызывает торможение катодного процесса (катодный контроль). Причинами появления пленки влаги являются: 1) капиллярная конденсация влаги – обусловлена зависимостью давления насыщенных паров от формы поверхности и степени кривизны мениска жидкости, над которым устанавливается равновесное давление паров Р. Над выпуклым мениском Р больше, чем над вогнутым (рис. 4.22.2). При этом зависимость Р от радиуса кривизны вогнутого мениска r определяется уравнением Томсона: где Р1 и Р0 – давление насыщенного пара над вогнутым и плоским мениском соответственно; σ – поверхностное натяжение жидкости; vm – объем моля жидкости; R – универсальная газовая постоянная; Т – абсолютная температура; r – радиус кривизны вогнутого мениска. С уменьшением r вогнутого мениска уменьшается давление насыщенных водяных паров над этим мениском. Наличие щелей и зазоров на поверхности металлоконструкций, представляющих собой капилляры со смачивающимися стенками, вызывает конденсацию водяного пара, не насыщенного по отношению к плоскому мениску жидкости. В щелях и зазорах возможна капиллярная конденсация и застой влаги, усиливающих атмосферную коррозию и вызывающих образование коррозионных язв. 2) Адсорбционная конденсация влаги обусловлена адсорбционными силами на поверхности металла. В широких капиллярах преобладает капиллярная конденсация, в тонких – эффект действия адсорбционного поля. 3) Химическая конденсация влаги– развитие адсорбционной конденсации в виде химического взаимодействия продуктов коррозии с водой с образованием гидратированных соединений с пониженным давлением насыщенных паров воды. Дальнейшую конденсацию влаги облегчают пленки растворов солей на поверхности металла, которая снижает давление насыщенного водяного пара. Для защиты от атмосферной коррозии применяют органические, неорганические и металлические покрытия, эффективны методы, воздействующие на процессы, контролирующие влажную атмосферную коррозию: а) торможение анодного процесса легированием стали легко пасивирующими металлами (хромом, алюминием, титаном, никелем) или эффективными катодными добавками, облегчающими пассивирование стали в атмосферных условиях; б) снижением влажности воздуха (осушкой), способствующим уменьшению толщины слоя электролита на поверхности металла, затруднением конденсации влаги (повышением температуры помещений) и уменьшением загрязненности воздуха. Применяют контактные, наносимые на поверхность стальных изделий ингибиторы (NaNO2) и летучие (нитриты, карбонаты и др.) ингибиторы, которые используют для защиты металлопродукции на этапе хранения и транспортировке. Морская коррозия Коррозия в естественных нейтральных водных средах (морская, речная, озерная вода) преимущественно протекает по электрохимическому механизму. Основными факторами, определяющими скорость коррозии металлов в морской воде, является концентрация растворенных солей, жесткость воды, температура, концентрация в ней кислорода и скорость потока. Присутствие эффективных ионов-активаторов Cl‾ определяет способность морской воды инициировать возникновение питтинговой коррозии металлических материалов в пассивном состоянии. Значительное ускорение морской коррозии вызывает обрастание растительными и животными микроорганизмами подводной части - биокоррозия. Наиболее эффективным способом борьбы с биокоррозией как в пресных, так и морских водах является нанесение лакокрасочных покрытий со специальными биоцидными добавками. Приливы, отливы вызывает усиленную коррозию по ватерлинии, что обусловлено облегченным доступом кислорода к поверхности металла. Испарение воды выше области смачивания вызывает увеличение концентрации солей в этой зоне. Удаление окалины со стального листа химическим травлением, пескоструйной обработкой или пламенем устраняет причины усиленной морской коррозии. Эффективным способом защиты сооружений и конструкций от морской коррозии является электрохимическая защита (протекторная или катодная) в комбинации с защитными покрытиями. Биокоррозия Биокоррозия - коррозия металла под влиянием жизнедеятельности микроорганизмов. Обычно протекает совместно с атмосферной или подземной коррозией в водных растворах или неэлектролитах. Причины ускорения коррозионных процессов связаны с жизнедеятельностью бактерий: 1) на поверхности металла образуются различные концентрационные элементы; 2) в растворе и на поверхности металла образуются агрессивные химические соединения являющиеся продуктами жизнедеятельности организмов; 3) меняются электрохимические свойства среды, что связано с изменением концентрации кислорода или других окислителей в растворе. Биокоррозию подразделяют на бактериальную и микологическую (грибную). Бактериальная коррозия протекает в водных средах при температурах 6 - 40оС и рН=1 - 10, 5 в присутствии органических и неорганических веществ. Микроорганизмы, использующие в качестве источника энергии неорганические вещества образуют группу литотрофных бактерий: 1)водородные, окисляющие водород с образованием воды; 2) нитрифицирующие, окисляющие аммиак до азотной кислоты; 3) тионовые, окисляющие сероводород до элементарной серы или элементарную серу до серной кислоты; 4) железобактерии, окисляющие закисное железо до окисного; 5) метанобразующие, стимулирующие природный синтез метана из углекислоты и водорода в анаэробных условиях (безкислородная среда); 6) сульфатвосстанавливающие, жизнедеятельность которых происходит за счет процесса восстановления сульфатов до сероводорода; 7) нитратвосстанавливающие, вызывающие восстановление окисленных форм азота по схеме: нитрат – нитрит – азот – аммиак. Наиболее опасными являются тионобактерии и железобактерии. Первые окисляют серу до серной кислоты, локальная концентрация которой в приповерхностном слое раствора может достигать 10%. Вторые поглощают железо в ионном состоянии и выделяют его в виде нерастворимых соединений, неравномерно распределенных по поверхности металла, что повышает электрохимическую гетерогенность поверхности и ускоряет коррозионные процессы. Основной вид коррозионно-активных анаэробных бактерий – сульфатвосстанавливающие. В процессе жизнедеятельности они восстанавливают сульфат-ионы в сульфит-ионы: Микологическая (грибная) коррозия – разрушение металлов и металлических покрытий при воздействии агрессивных сред, формирующихся в результате жизнедеятельности микроскопических (несовершенных, плесневых) грибов. Грибы непосредственно коррозию не вызывают, основная форма влияния плесневых грибов - косвенное воздействие через продукты метаболизма гриба. Причина повышения скорости коррозии в присутствии грибов – облегчение активирования защитных и пассивных пленок. Защита металлов от биокоррозии в основном состоит в предотвращении, ограничении или уничтожении микроорганизмов, что достигается: - повышением общей коррозионной стойкости металлов и покрытий; - применением лакокрасочных покрытий и полимерных материалов, обладающих биоцидными свойствами, или включающих биоциды; - нанесением на поверхность конструкций смесей, включающих гидрофобизирующие, ингибирующие вещества и биоциды; - поддержанием определенных условий эксплуатации (относительная влажность воздуха < 80 %, температура < 20оС, воздухообмен, очистка воздуха и поверхностей от механических загрязнений); - вводом в водные среды эффективных добавок бактерицидов; - применением катодной и протекторной защиты для подземных сооружений, гидросооружений и плавучих средств; - применением средств консервации, содержащих ингибиторы коррозии, в том числе летучие. Локальные виды коррозии Межкристаллитная коррозия (МКК) - преимущественное разрушение границ зерен поликристаллического металла при пренебрежимо малой скорости общей коррозии. МКК в той или иной степени подвержены коррозионностойкие стали всех структурных классов – ферритные, мартенситные, аустенитно-ферритные, аустенитные, а также дисперсионно-твердеющие алюминиевые сплавы. Условия возникновения МКК сталей и алюминиевых сплавов различны, но характер проявления практически одинаков: при общей высокой коррозионной стойкости происходит избирательное растворение границ зерен. Причины МКК: - высокое содержание углерода и отсутствие стабилизирующих элементов (для хромонокелевых сталей); - неправильно проведенная термообработка; - технологические операции в опасном температурном интервале (сварка, штамповка, ковка, волочение и др.); - длительная эксплуатация при повышенной температуре; - неправильный выбор структурного класса для эксплуатации в определенной коррозионной среде. В сварных соединениях МКК может возникать: а) в основном металле, в зоне термического влияния, где металл подвержен нагреву в области опасных температур; б) на границе наплавленного металла и основного металла – в виде ножевой коррозии; в) в наплавленном металле. МКК коррозионностойких сталей наблюдается обычно после термообработки, приводящей к образованию на границах зерен каких-либо новых фаз, чаще всего карбидов. Их причиной образования является различная зависимость растворимости углерода от температуры в фазах. В хромистых ферритных сталях растворимость углерода низка, поэтому в них карбиды по границам зерен выпадают уже в процессе охлаждения от высоких температур. Отжиг ферритных сталей при 600-800оС приводит к исчезновению склонности к МКК из-за коагуляции выделений карбидов. В сталях аустенитного и аустенитно-ферритного класса углерод имеет относительно более высокую растворимость и закалка позволяет зафиксировать состояние твердого раствора углерода, поэтому карбиды не образуются. Их образование происходит только при отпуске или замедленном охлаждении. Теории МКК. 1. Теория обеднения - наиболее распространенная и экспериментально подтвержденная теория, объясняющая МКК в слабоокислительных средах. Основная причина МКК – обеднение границ зерен аустенита хромом, находящимся в состоянии твердого раствора, в результате образования по границам зерен при отпуске закаленных сталей богатых хромом фаз, таких как (Fe, Cr)23C6 или Cr3C. Уменьшение концентрации хрома в области границы зерна ниже критической, обеспечивающей возможность пассивирования, приводит к образованию локального гальванического элемента, в котором пассивное тело зерна является катодом, а периферийные участки (граница зерна), находящиеся в активном состоянии – анодом. При более высокой скорости диффузии углерода, по сравнению со скоростью диффузии хрома, необходимый для образования карбида углерод поставляется с границ зерен, из прилегающих областей, и из тела зерна. Хром в начале процесса образования Cr3C поступает с границ или прилегающих участков, поэтому диффузия не успевает восполнять израсходованное количество хрома. Но чем выше температура, тем выше скорость диффузии хрома, поэтому, чем выше температура отжига, тем ниже склонность к МКК. Теория химически нестойкой фазы основана на фактах разрушения границ зерен в окислительных и сильно окислительных средах (HNO3). Наблюдается при образовании фаз, содержащих молибден (карбидов, σ -фаза и др.). Теория микроэлементов- при контакте коррозионной среды и коррозионностойкой стали, имеющей выделения карбидов по границам зерен, образуется микроэлемент. Он локализуется около карбида (катода), а обедненные хромом прилегающие к нему участки – аноды – сильно корродируют. Развитие МКК по этому механизму связано с образованием сплошных или слабо разобщенных карбидных выделений. Теория напряжений - напряжения, возникающие при образовании и росте карбидной фазы на границах зерен приводят к возникновению разности потенциалов, облегчению протекания анодного процесса. Теория сегрегаций - образовании сегрегаций углерода и хрома по границам зерен в рамках теории обеднения при кратковременных нагревах. В области перехода из активного состояние в пассивное наиболее достоверен механизм обеднения границ; в пассивной области ─ механизм обеднения; в транспассивной области преобладает сегрегационный механизм. Способы борьбы с МКК нержавеющих сталей: 1) снижение содержания углерода до предела его растворимости при соотвествующих температурах; 2) закалка или перезакалка с целью перевода хрома и углерода в состояние твердого раствора; 3) легирование стали карбидообразующими элементами ─ титаном, ниобием, танталом, которые связывают углерод в соответствующие карбиды, где хром находится в состоянии твердого раствора; 4) длительный нагрев (более двух часов) сталей при достаточно высоких температурах (550 - 900оС) с целью коагуляции карбидов; 5) аустенизация. Для алюминиевых сплавов термическая обработка (искусственное старение), проводящаяся для упрочнения (дисперсионное твердение) вызывает понижение коррозионной стойкости. Склонность к МКК магналиев (сплавов алюминия с 5 - 10% Mg) можно устранить отпуском при 250 - 400оС. В указанной области температур происходит коагуляция анодных частиц Mg2Al3, что нарушает непрерывность цепочки выделений этой фазы. Питтинговая коррозия (ПК)- точечная коррозия (местная коррозия металла в виде отдельных точечных поражений) проявляется на металлах, находящихся в пассивном состоянии, когда коррозии с высокой скоростью подвергаются отдельные небольшие участки поверхности, что приводит к образованию глубоких поражений – точечных язв, или питтингов. ПК проявляется в окислительных средах в присутствии активирующих галогенид-ионов (Cl‾, Br‾, J‾ ), сульфат-иона ( Механизмы питтинговой коррозии: а) механизм проникновения аниона – миграция анионов через оксидную пленку к границе раздела МеО/Ме под влиянием электрического поля напряженностью 1мВ/см2; б) механическое разрушение пассивной пленки, которое вызывается проникновением электролита через ослабленный участок поверхности металла, при этом агрессивные ионы предотвращает репассивацию поверхности. Разрушение пленки происходит под влиянием электрострикции, вызванной адсорбцией анионов Cl‾; и) адсорбционный механизм – адсорбция активирующих анионов на определенных участках поверхности пассивирующего оксида. Адсорбированные анионы образуют поверхностные комплексы с металлическими ионами, вытесняя кислород из решетки оксида. Это облегчает переход ионов металла в электролит и вызывает уменьшение толщины оксидной пленки, которое сопровождается увеличением напряженности электрического поля, и, следовательно, увеличением скорости миграции металлических ионов. Растворение металла в питтинге имеет электрохимическую природу - поверхность питтинга является анодом, поверхность металла с пассивной пленкой МеmОn – катодом. Развитие питтинга идет в несколько стадий: 1) нарушение пассивности; 2) возникновение питтинга; 3) начальный этап роста питтинга (характеризуется непрерывным ростом тока и появлением первых питтингов размером несколько микрометров); 4) репассивация, или прекращение роста питтинга наступает (если возможно) после второй стадии и характеризуется возвратом системы в пассивное состояние, питтинг зарастает; 5) стабильный рост питтинга. На стадии 2) возникновения питтинга растворяются структурные элементы поверхности металла под участками с менее совершенной пассивацией, имеющими более отрицательное значение Епо, чем остальные участки запассивированной поверхности. Несовершенства поверхности связаны с искажениями структуры: границы зерен, выходы дислокаций, металлические и неметаллические включения, выходом на поверхность металла кристаллитов с менее благоприятной для пассивации ориентацией, сегрегациями примесей. Питтинг на этом этапе представляет собой ямку от растворившегося кристалла. Если образование предпиттинга связано с менее благоприятными для пассивации структурными включениями, но изменений концентрации коррозионной среды, то предпиттинг репассивирует. Но если после растворения дефектного участка в зародыше питтинга повышается активность среды (Cl‾, H+), то развитие питтинга перейдет во вторую фазу, когда по мере протекания коррозионного процесса в питтинге агрессивность среды сильно возрастает, и среда подкисляется вследствие гидролиза растворяющихся ионов металла: Ме→ Меn++ne; Меn++H2O→ Me(OH)(n-1)+ +H+. Особенностью стадии 3, является возможность репассивации, которая может произойти вследствие смещения потенциала металла в более отрицательную область потенциалов, в область устойчивой пассивности. Причиной репассивации может являться также переход ионов легирующих добавок в металле (Mo, Si, W, Re, Ru) в виде оксоанионов ( В отсутствие репассивации начинается стабильный рост питтинга. Происходит дальнейшее повышение агрессивности среды. Возрастание потенциала в питтинге, вызванное ограничением доступа электролита по мере увеличения глубины питтинга, будет несколько снижать скорость его роста, однако репассивация не произойдет – установиться стабильный режим роста питтинга (стадия 5). Механизм и склонность к питтинговой коррозии обычно оценивается по анодным поляризациолнным кривым (рис.4.23).
Рис. 4.23. Обобщенная анодная поляризационная кривая: Енп – потенциал начала пассивации; Ереп – потенциала репассивации; Епо- потенциал питтингообразования; Епр- потенциал перепассивации; потенциал питтингообразования (Епо) – минимальное значение потенциала, при котором возникает ПК, характеризуется устойчивым увеличением тока на поляризационной кривой при достижении потенциала Е> Епо; потенциал репассивации (Ереп) – потенциал, при котором прекращается работа питтингов, возникающих при Е> Епо. Методы защиты от питтиговой коррозии. 1. Выбор сплава - устойчивы Сг-стали. Более высокой стойкостью к ПК отличаются высокохромистые ферритные стали повышенной чистоты по примесям внедрения, содержащие 18 или 25 - 29 % Сг, а также эти стали, дополнительно легированные 1 - 4 % Мо. Из аустенитных сталей наиболее стойки к ПК стали, легированные 2 - 3 % Мо, - Х18Н12М2Т и Х18Н12МЗТ, а также с более высоким содержанием Мо, например 03Х18АГЗН12М5. Стали Х20Н20, содержащие > 3 % Si, также отличаются повышенной стойкостью к ПК. В очень агрессивных условиях: (при повышении концентрации галоидных анионов и температуры) следует использовать Ti или его сплавы Ti - (0, 15 + 0, 20) % Pd, TI - 2, 5 % Ni - 2 % Zr, Ti - 2 % Ni - (1 + 2) % Mo -наиболее стойкие к ПК из доступных конструкционных материалов. 2. Электрохимическая защита и применение ингибиторов. Для предотвращения ПК можно сместить потенциал сплава или в сторону менее положительных значений пассивной области в область Епо - Епп (анодная), или отрицательнее стационарного потенциала Ест (катодная). Надежность применения анодной защиты сталей от ПК повышается в случае присутствия ингибиторов в растворе - NaN03, гипофосфита натрия, нитратов, хроматов, сульфатов, щелочей и органических ингибиторов.
Популярное:
|
Последнее изменение этой страницы: 2017-03-09; Просмотров: 949; Нарушение авторского права страницы
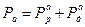 ,
, 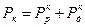 . Для контролирующего процесса коррозии металла желательно установить его элементарную контролирующую стадию – электродную реакцию или диффузию, сопоставляя Pa и Рк. Это делается на основании взятых из коррозионной диаграммы (расчетов), величин коррозионного тока I1 (точка В, рис. 4.18) и
. Для контролирующего процесса коррозии металла желательно установить его элементарную контролирующую стадию – электродную реакцию или диффузию, сопоставляя Pa и Рк. Это делается на основании взятых из коррозионной диаграммы (расчетов), величин коррозионного тока I1 (точка В, рис. 4.18) и
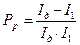 ,
, 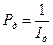 . (4.87)
. (4.87) или
или 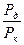 будет характеристикой элементарной контролирующей стадии контролирующего процесса.
будет характеристикой элементарной контролирующей стадии контролирующего процесса.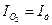 и процессом выделения водорода
и процессом выделения водорода 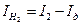 :
:  ; (4.88)
; (4.88) , (4.89)
, (4.89)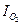 , и
, и 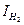 для определения степени контроля определяются по поляризационной коррозионной диаграммы или из расчетов.
для определения степени контроля определяются по поляризационной коррозионной диаграммы или из расчетов.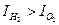 ). Наблюдается при коррозии железа, цинка и других металлов в растворах неокисляющих кислот при низком pH, а также при коррозии магния и его сплавов в нейтральных растворах из-за отрицательного значения (
). Наблюдается при коррозии железа, цинка и других металлов в растворах неокисляющих кислот при низком pH, а также при коррозии магния и его сплавов в нейтральных растворах из-за отрицательного значения ( 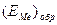 ;
; 

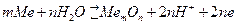 . (4.90)
. (4.90)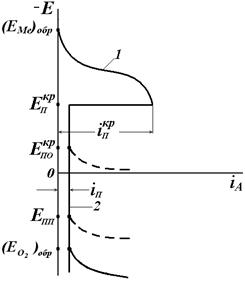
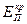 – критический потенциал пассивации,
– критический потенциал пассивации, 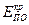 – критический потенциал питтингообразования,
– критический потенциал питтингообразования, 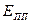 – потенциал перепассивации,
– потенциал перепассивации, 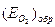 – обратимый потенциал выделения О2. Характерные токи:
– обратимый потенциал выделения О2. Характерные токи: 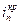 – критический ток пассивации,
– критический ток пассивации, 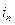 – ток пассивного состояния; б – схема поляризационной коррозионной диаграммы, поясняющая самопассивацию Fe в HNO3.
– ток пассивного состояния; б – схема поляризационной коррозионной диаграммы, поясняющая самопассивацию Fe в HNO3.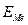 в отсутствии внешнего тока равен
в отсутствии внешнего тока равен 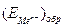 . На кривой выделяется область активного растворения металла (1) и область пассивности (2). Потенциал активно-пассивного перехода применительно к обобщенной кривой называется критическим потенциалом пассивации
. На кривой выделяется область активного растворения металла (1) и область пассивности (2). Потенциал активно-пассивного перехода применительно к обобщенной кривой называется критическим потенциалом пассивации 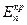 , а соответствующий ему максимальный ток активного растворения – критическим током пассивации
, а соответствующий ему максимальный ток активного растворения – критическим током пассивации 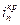 . Начальный участок кривой 1 описывается тафелевской зависимостью, и дальнейшие отклонения от неё связаны с началом процесса пассивации. В области 2 величина тока пассивации iп не зависит от потенциала. Для некоторых оксидов металлов с высокой электронной проводимостью (Fe, Ni) при увеличении потенциала до обратимого потенциала кислородного электрода
. Начальный участок кривой 1 описывается тафелевской зависимостью, и дальнейшие отклонения от неё связаны с началом процесса пассивации. В области 2 величина тока пассивации iп не зависит от потенциала. Для некоторых оксидов металлов с высокой электронной проводимостью (Fe, Ni) при увеличении потенциала до обратимого потенциала кислородного электрода 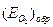 возможна реакция выделения кислорода. На хроме при более отрицательном потенциале, чем
возможна реакция выделения кислорода. На хроме при более отрицательном потенциале, чем 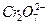 или
или 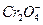 анионов. Этот процесс называемся перепассивацией. Для хрома перепассивация имеет важное практическое значение для коррозии хромистых и хромоникелевых сталей в сильных окислителях.
анионов. Этот процесс называемся перепассивацией. Для хрома перепассивация имеет важное практическое значение для коррозии хромистых и хромоникелевых сталей в сильных окислителях.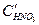 . С увеличением концентрации
. С увеличением концентрации 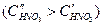 катодная кривая К2 пересекает анодную в пассивной области, так что скорость коррозии металла
катодная кривая К2 пересекает анодную в пассивной области, так что скорость коррозии металла 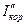 равна току пассивности
равна току пассивности 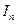 . Различие в ходе катодных кривых К1 и К2 объясняется скоростью протекания реакции:
. Различие в ходе катодных кривых К1 и К2 объясняется скоростью протекания реакции: 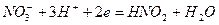 . (4.91)
. (4.91)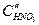 , скорость реакции выше, торможение реакции и перенапряжение катода меньше. Коррозия Fe здесь начинается с активного растворения Fe, и, когда ток растворения превосходит критический ток пассивности
, скорость реакции выше, торможение реакции и перенапряжение катода меньше. Коррозия Fe здесь начинается с активного растворения Fe, и, когда ток растворения превосходит критический ток пассивности 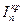 , металл переходит в пассивное состояние. При достаточно низких концентрациях кислоты коррозия Fe будет протекать с высокой скоростью, а при достаточно высоких концентрациях железо пассивируется. При очень высоких
, металл переходит в пассивное состояние. При достаточно низких концентрациях кислоты коррозия Fe будет протекать с высокой скоростью, а при достаточно высоких концентрациях железо пассивируется. При очень высоких  снова возможен рост в области перепассивации Fe. Эта зависимость скорости роста коррозии Fe от концентрации HNO3 хорошо объясняет экспериментальные данные (рис. 4.20).
снова возможен рост в области перепассивации Fe. Эта зависимость скорости роста коррозии Fe от концентрации HNO3 хорошо объясняет экспериментальные данные (рис. 4.20).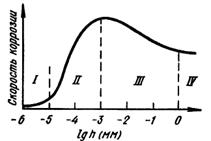
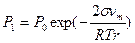 , (4.92)
, (4.92) .
. ), в смесях соляной и азотной кислот, в морской воде, в хлоридных растворах железа, хлористого натрия и др.
), в смесях соляной и азотной кислот, в морской воде, в хлоридных растворах железа, хлористого натрия и др.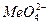 ), которые осаждаются на его поверхности, вытесняя ионы Cl‾. Это вызывает торможение анодного процесса в питтинге и репассивацию последнего.
), которые осаждаются на его поверхности, вытесняя ионы Cl‾. Это вызывает торможение анодного процесса в питтинге и репассивацию последнего.