|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Современные методы картирования геномов.
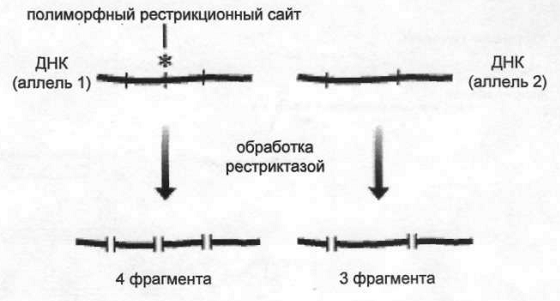
Картирование – самый простой способ состыковать контиги. Картирование геномов может быть генетическое и физическое. Генетическое картирование: Содержит ген – фенотипический маркер; Молекулярные маркеры: RFLP (полиморфизм длин рестрикиционных фрагментов) –это способ исследования геномной ДНК, путем разрезания ДНК с помощью эндонуклеаз рестрикций (расщепляют нуклеиновые кислоты в середине) и дальнейший анализ образовавшихся фрагментов (рестриктов) путем гель-электрофореза; SSLP (полиморфизм длин простых повторов)- Их есть 2 типа: минисателлиты и микросателлиты. Микросателлиты – повтор 2, 3, 4 н.п. Это ошибки в работе ДНК-полимеразы. Когда есть участок, где много раз повторяется олигонуклеотид, есть вероятность проскальзывания ДНК-полимеразы. Она с определенной долей вероятности может диссоциировать с матрицей и присоединиться к соседнему повтору. В результате при репликации будет либо на один повтор больше, либо на один меньше; SNP (Полиморфизм по одиночным нуклеотидам) – отличия последовательности ДНК размеров в 1 нуклеотид в геноме представителя одного вида; Детекция молекулярных маркеров: 1. Гибридизация ДНК: 1.1. Зонды будут спариваться с одноцепочечной ДНК, образуются полные или неполные комплементы. 1.2. Электрофорез в ПАА. Перенос с помощью нитроцеллюлозной бумаги. Радиомеченные пробы гибридизуются с ДНК. Выявляются авторадиографически. 2. ПЦР:
Используется для детекции молекулярных маркеров: 1. Повторяющихся локусов;
2. Полиморфизм рестрикционных фрагментов (ПДРФ, RFLP) – обработка рестриктазой.
Типы повторов: 1. Короткие (2-4 н.) полиморфизм длин простых последовательностей (SSLP);
2. Минисателлиты ( до 25 н.) VNTR; 3. Микросателлиты (ди- или тетрануклеотиды) STR, SSR; Полиморфизм по одиночным нуклеотидам в агарозном геле (SNP); (AGTCA G AAATC); (AGTCA C AAATC);
Использование ДНК-чипов методом фотолитоавтографии. Стеклянная подложка обрабатывается, на неё наносится нуклеотид. 5’-конец блокируется, освещение лазером удаляет блокирующую группировку. Так можно нанести сотни тысяч зондов.
Негибридизующиеся фрагменты отмываются; определяется по пятнам (от флуоресцентно меченной ДНК).
Недостатки: 1. Ограниченная разрешающая способность (максимум 1 ген на 3.3 кн. у E. coli и 1 ген на 10 кн. У S. cerevisiae); 2. Ограниченная разрешающая способность из-за неравномерности кроссинговера.
Основные методы физического картирования: 1. Рестрикционное картирование; 2. FiSH (флуоресцентная гибридизация in situ); 3. Картирование STS;
Рестрикционное картирование – это определение относительного расположения и определенных сайтов рестрикции на геномной карте и определение расстояний между ними. Особенности крупных участков: 1. Использование рестриктаз, распознающих 7-8 н.; 2. Использование рестриктаз, в сайт распознавания которых входят редкие комбинации нуклеотидов; 3. Использование специальных методик гель-электрофореза с переменным электрическим полем для разделения крупных фрагментов ДНК. Классическим походом является электрофорез для определения размеров фрагментов. Нужно как минимум 2 рестриктазы, каждая из них обрабатывает исследуемый фрагмент ДНК. Первое что нужно сделать это подобрать рекстриктазу которая разрезает реже, чтоб получилось разумное число фрагментов. Здесь вот статистически рассчитанная частота встречаемости сайтов для различных эндонуклеаз с разным размером сайта. Берутся рестриктазы которые имеют самый длинный сайт, плюс подбираются специально еще такие рестриктазы в сайтах которых есть редкие комбинации нуклеотидов. Таким образом мы получаем реакцию рестрикции с разумным количеством фрагментов, но нужно все равно определить размер этих фрагментов. Для того чтобы определить размер крупных фрагментов используются специальные методики: либо электрофорез в переменном поле, либо другие способы визуализации которые объединяются под названием оптическое картирование. Первое – таким образом можно найти можно найти ферменты, которые будут еще реже резать. В частности на примере генома человека, этот сайт показывает почему у нас мало CG комбинаций. В геноме человека CG буквы расположены рядом встречаются только в промоторных областях. Соответственно в большей части генома они не присутствуют. Почему так происходит? Потому что цитозин метилируется, т.е. происходит присоединение метильной группы. Был цитозин, получился 5-метилцитозин. Любая аминогруппа в составе азотистых оснований с достаточно высокой вероятностью способна гидролизоваться. Остаток от гидролиза – замена аминогруппы на оксогруппу, а если происходит замена цитозина, то мы получаем урацил который является четным основанием и соответственно системой репарации вырезается, но если это 5-метилцитозин, то мы получаем тимин, а это легальное основание в составе ДНК. Соответственно если вот буква С не критична в соответствующем месте геномной последовательности, то эволюционное давление достаточно жесткое - замена буквы С на букву Т. Эволюционное давление будет больше в кодирующей последовательности и в промоторной последовательности, потому что метилированные CG последовательности являются важным регуляторным сигналом который контролирует. Это так называемые CPGS-права там где они есть почти наверняка будет регуляторная область, которая активирована. Таким образом если задача – подобрать редкощепящие рестриктазы, то потребуется подобрать такие рестриктазы в сайт которых входят эти комбинации нуклеотиды. Давайте посмотрим что получается. Один из вариантов это электрофорез в пульсирующем поле. Стандартный электрофорез очень плохо подходит для фрагментов с более 50 тыс. н.п. Они все собираются в кучку наверху геля, не разделяются, некоторые из них вообще остаются в лунке. Чтобы их разделить меняются условия электрофореза – два электрода меняются на 4 и напряжение подается переменное то на один электрод, то на другой. Когда подается напряжение на первый электрод фрагмент ДНК движется очень короткое расстояние, застревает в порах агарозы. Напряжение меняется, ток подается на другие электроды, фрагмент ДНК выдергивается из тех ячеек в которых он застрял и чуть-чуть двигается, потом направление опять меняется. Движение получается зигзагообразное. Но шажки очень короткие и суммируются в линейное движение. Фактически получается обычный электрофорез, разница в том что крупные фрагменты будут четко разделены, т.к. подвижность совсем другая. Не так жестко привязана к массе фрагментов. У электрофореза есть один большой недостаток – это не прямое измерение размеров ДНК. По одной из дорожек будет бежать молекулярный маркер и идет сопоставление длины пробега – косвенный показатель.
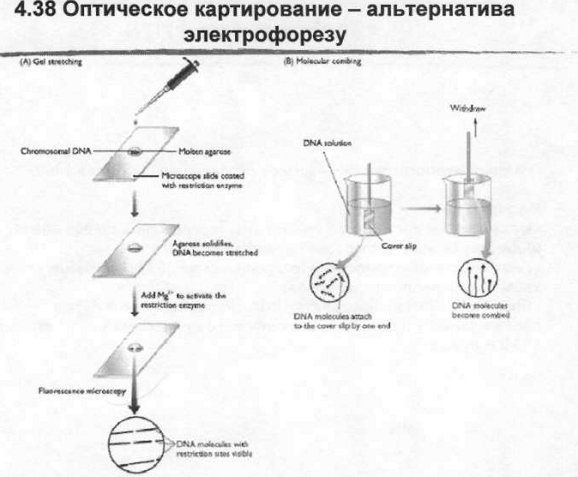
То, что называется оптическим картированием – это замена электрофорезу, здесь размер фрагментов измеряется непосредственно. Суть его заключается в том, что ДНК одним из нескольких способов распрямляется, из скрученного клубка она вытягивается. Причем вытягивается под напряжением. Причем напряжение молекулы ДНК, обрабатываются рестриктазой, после разрезания концы молекулы разъезжаются в стороны от места разреза это все можно увидеть под микроскопом. Потом такой препарат микроскопируется, ДНК окрашивается интеркалирующим флуоресцентным красителем и места разрыва видны. Прямое измерение длины под микроскопом. Два варианта получения линейные напряженные ДНК: в обоих случаях используется свойство границы между различными фазами. Когда ДНК проходит через фазовый раздел вытягивается. Первая из методик использует ДНК в растворе расплавленной агарозы. ДНК наносится на предметное стекло, через некоторое время агароза начинает застывать. Застывание никогда не идет равномерно, оно всегда начинается в каком-то месте. Фронт кристаллизации агарозы движется с начальной точки и распространяется. Когда он проходит через молекулу ДНК он тянет эту молекулу за собой и в результате мы получаем выровненную молекулу в напряженном состоянии. Потом сюда добавляется фермент, в некоторых случаях фермент сразу иммобилизован на стекле, а потом добавляются ионы магния вместе с соответствующим буфером, а может быть наоборот – потом фермент. Но лучше первый вариант – лучше диффундирует. Другой метод основан на том что покровное стеклышко очень медленно достается из раствора ДНК. Оно обрабатывается раствором силаном который увеличивает сорбцию ДНК, молекулы какой-то частью сорбируются на этом стеклышке и оно очень медленно, по паре миллиметров в час вытаскивается из раствора и когда оно проходит через пленку поверхностного натяжения молекулы ДНК вытягиваются от того места где они зафиксированы вниз. Направление противоположное движению стеклышка. Мы получаем выровненную молекулу в напряженном состоянии, потом стеклышко обрабатывается рестриктазой.
При неспаренном состоянии гибридизации нет. Детекция SNP методом гибридизации в растворе. При гибридизации шпилька раскрывается. Следующая методика картирования флуоресцентная гибридизация на препаратах – FISH. Здесь непосредственно визуализуется какой-нибудь локус, в соответствии с позицией флуоресцентного зонда на окрашенных цитологическим красителем препарата хромосом (скрученные хромосомы). Препарат наносится на стекло, фиксируется на нем и подвергается частичной денатурации. В результате мы получаем по длине хромосомы участки в одноцепочечном состоянии. Затее добавляется проба которая гибридизуется с этими участками, и затем такой аппарат окрашивается соответствующими красителями и микроскопируется. Микроскопия ведется в видимом диапазоне, микроскопия флуоресцентная. Проба облучается лазером в определенной длине волны и наблюдается флуоресценция. В результате мы поучаем картину флуоресценции наложенную на характерную для данной хромосомы картину полос. Мы сделали зонд под конкретный ген и сразу видим где на хромосоме этот ген расположен. Недостатки: разрешающая способность достаточно невысокая, не более 1 млн.н. п. между соседними зонами. Чтобы повысить разрешение используются модификации методик. Самая простая фактически ничего не меняет – механическое растяжение хромосом. Препарат хромосом крутится в центрифуге на небольших оборотах. Хромосомы вытягиваются под действием центробежных сил, главное не переборщить с оборотами. Белки не соединяются остаются в тех позициях в которых они зафиксированы и можно окрасить. Разрешение в 4-5 увеличивается. До 200 тыс.н.п. между соседними маркерами. Если нужно более высокое разрешение используются другие модификации – не метафазные хромосомы (разрешение до 20-25 тыс.н.п.) и fiber-FISH, те же самые хромосомы, только которые дополнительно растянуты – растягивание в геле, молекулярное «причесывание», разрешение до 10 тыс. н. п. Недостаток этих двух методик – мы не можем привязать флуоресцентный зонд к цитологической карте. Мы можем только расположить 2 зонда друг относительно друга и определить расстояние между зондами. Должен быть зонд позиция которого известна, и мы должны одновременно видеть и этот зонд и тот зонд который нас интересует.
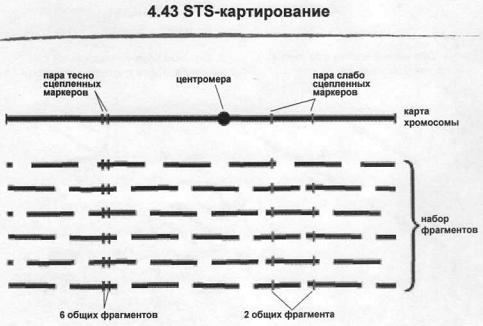
Еще один способ картирования это STS-картирование. Оно чем-то похоже на генетическое картирование, но использует не рекомбинацию, а другой способ определения относительного расстояния. Критерием здесь служит частота фрагментации ДНК при построении геномных библиотек и фрагментов. Фактически суть та же самая: разрыв молекулы ДНК происходит под действием низкого ультразвука или под действием рестриктаз. отличие также в том, что служит маркерами. STS-маркеры – это известные фрагменты ДНК, которые встречаются в геноме один раз и последовательность должна быть известна. Когда такая последовательность известна можно подобрать пару праймеров в пределе этих 100-500 н. п. и получить ПЦР фрагмент, по которому обычно такой маркер и детектируется. Самый простой способ отбора последовательностей для получения этих маркеров – это использовать EST сиквенсы это коротенькие кусочки кДНК, сделанные на матричной РНК. Поскольку это копия мРНК это она гарантированно встречается один раз в геноме, поскольку большинство генов является уникальными. Соответственно это изначально уникальная последовательность. Другие маркеры, менее удобные, маркеры полиморфные по простым последовательностям – минисателлиты или микросателлиты. Можно использовать и случайные геномные последовательности, но с ними сложнее т.к. нужно четко доказывать что они уникальные геномные последовательности. Для того, чтобы определить позицию таких маркеров нужен другой компонент – реагент картирования. Это просто набор фрагментов ДНК той или иной библиотеки. Чем маркеры ближе друг к другу на реальной карте хромосомы, тем выше вероятность, что они окажутся в одном и том же фрагменте ДНК. Тесно сцепленные маркеры, рядом расположены соответственно они попадают в большое число фрагментов. Маркеры расположенные дальше, чаще всего попадают на разные фрагменты ДНК, Если маркеры окажутся еще дальше – расстояние между ними будет превышать размер фрагментов библиотеки, они вообще не будут детектироваться совместно. Все это статистически общипывается – можно по частоте совместного нахождения этих маркеров в библиотеке достаточно точно определить расстояние между ними на реальной карте хромосом. При условии что фрагментация лишь случайна. Источники фрагментов: стандартная клонотека, набор клеток-радиационных гибридов.
Клетки-радиационные гибриды наиболее удобны при STS-картировании. Это старая технология, фактически вариант клонирования эукариотической ДНК in vivo. Речь идет о гибридизации облученных клеток с необлученными клетками. Причем необлученные клетки, как правило, клетки китайского хомячка, а облученные – клетки любого млекопитающего в т.ч. человека. Если допустить, что это человеческие клетки, они подвергаются воздействию очень высоких доз γ -радиации и такая клетка становится нежизнеспособной, мы получаем клетку с серьезно фрагментированной ДНК и репарировать такие хромосомы невозможно. Если такую клетку слить с клеткой другого млекопитающего, то система репарации неповрежденной клетки будет пытаться репарировать эти двунитевые разрывы и результатом будет случайные включения фрагментов вот этой ДНК в различные участки своих неповрежденных хромосом. То что не удалось включить при делении клетки будет элиминировано, а то что включилось будет стандартно наследоваться в этой клеточной линии. Поскольку в каждую хромосому включается несколько фрагментов чужеродной ДНК при правильной дозе радиации достаточно около 100 клеток ста линий таких гибридом для того, чтобы построить подробную STS-карту эукариотического генома. При помощи ПЦР зафиксируется наличие маркеров в одном образце ДНК и высчитывается частота нахождения этих маркеров во всех вариантах линий. Есть различные механизмы автоматизации. Суть его сводится к тому, что для детекции используется не электрофорез, а флуоресцентная метка, причем эта метка достаточно хитро сюда включается. Флуоресцентные нуклеотиды добавляются в реакцию, но система детекции способна улавливать только поляризованный сигнал, изначально сигнал не поляризован – детекция не идет. Ситуация включается при включении такого нуклеотида в состав ДНК. Эти нуклеотиды являются терминирующими поэтому они включаются только один раз и после включения флуоресцируют немного по-другому. Делается ПЦР, если продукт присутствует добавляется еще один внутренний праймер; он гибридизуется с одной из цепочек продукта и к нему присоединяется этот нуклеотид. После этого сигнал оказывается поляризованным и его можно детектировать. Если продукта нет – сигнала не будет. С такой модификацией ПЦР детекции можно в масштабах эукариотического генома достаточно быстро построить физическую карту генома. Когда мы начали разговаривать про sts-картирование, я говорил, что второй вариант источника фрагментов для картирования только стандартная клонотека. Она, немножко менее удобна в том плане, что гораздо больше клонов приходится использовать, ниже получается частота совместного нахождения на одном фрагменте, но скрининг получается более протяженный. Но, есть преимущества, которые по крайней мере частично компенсируют этот недостаток, они заключается в том, что одни и те же клоны можно использовать и для построения карты и те же самые клоны можно затем брать для секвенирования. Одну и ту же клонотеку можно использовать для картирования, а потом для определения нуклеотидной последовательности, поэтому тут не надо специально что-то конструировать, а уже имеющуюся клонотеку можно использовать для такого картирования.
Ну вам может быть рассказывали когда была цитология, про принцип fluorescence assisted cell sorting, то есть окрашивание этих клеток идет флуоресцентным красителем, то есть очень небольшая модификация этой методики позволяет открывать не клетки, а хромосомы. И этот принцип соответственно позволяет разделить весь хромосомный набор на отдельные хромосомы и затем, библиотеки делаются из ДНК одной хромосомы. Это колоссальная помощь потому что, одно дело работать в масштабе генома размером 3 млрд нуклеотидных пар, другое дело в масштабе одной хромосомы, тогда у нас получится в среднем 150 млн. Вы чувствуете, как падают масштабы и естественно этот метод используется. Я надеюсь, что вы это хорошо помните, здесь все предельно просто. То, что нужно сортировать, окрашивается флуоресцентным красителем, в данном случае хромосомы. Количество красителя, который связывается с хромосомой зависит от нескольких факторов, в первую очередь от размера хромосомы – это основной фактор, ну и во вторую очередь – это структура белков, которые связаны с этой хромосомой. Белки немножко различаются, в первую очередь различаются количеством гетерохроматина, ну это определяет то, что нет прямой корреляции между размером хромосомы и интенсивностью окрашивания. Ну, что происходит потом, потом раствор капается из пробирки маленькими каплями, каждая концентрация подобрана таким образом, чтобы капля была либо пустой, либо содержала одну хромосому, ни в коем случае не две. Затем капля сбоку освещается лучом лазера, и если в капле присутствует хромосома, то возбуждается флуоресценция, флуоресценция детектируется детектором. Интенсивность флуоресценции пропорциональна количеству красителя, который связался. Это количество характеризует каждую хромосому уникальным образом, вот на этой стадии можно сказать, какая хромосома в этой капле находится. Затем вот эта пара электродов в соответствии с интенсивностью флуоресценции заряжает каплю так, что получается достаточно мощное электрическое поле. Чем больше флуоресценция, тем больше заряд капле сообщается. Капля летит дальше, пройдя через следующую пару электродов. Эти электроды отклоняют каплю вправо или влево в соответствии с зарядом. Чем больше заряд, тем больше будет отклонение в одну сторону, в одну сторону она в общем будет отклоняться и соответственно каждая хромосома будет капать в свою пробирку. Правильный подбор красителя в случае хромосом человека позволяет таким образом раскапать все кроме одной пары хромосом по отдельным пробиркам. Для оставшейся пары хромосом берут другой краситель, который немного по другому связывается и эту пару тоже можно разделить. Таким образом, мы получаем из тотального препарата ДНК препараты отдельных хромосом, что существенно упрощает дальнейшую работу. На этом мы с картированием и библиотеками заканчиваем. Генетическое картирование до сих пор используется и будет использоваться т.к. помимо составления карт генома у него есть важное применение – есть методика маркерсистированной селекции которая позволяет очень быстро вводить нужный ген. Генотип такого нового сорт из дикого четко контролирует присутствие этого гена по молекулярному маркеру. Недостаток заключается в ограниченной точности. для E.coli и дрожжей генетическое картирование шло с 50х гг и была картирована тысяча генов. В случае E.coli и дрожжей максимальная плотность таких карт один ген на 3тыс.н.п. у E.coli и один ген на10 тыс.н.п. у дрожжей. И в том и в другом случае получается что удается картировать только каждый третий, каждый четвертый ген. Большей точности достигнуть не получается. Вторая проблема связана с тем что кроссинговер идет неравномерно по длине хромосомы, есть горячие точки кроссинговера, есть участки хромосомы которые блокируют кроссинговер – это вносит дополнительную ошибку и кроме того может поменять позиции некоторых локусов на генетической карте. Также из-за неаккуратности увеличивается число экспериментальных ошибок. У картирования 2 задачи – определить относительное расположение каких-то участков на хромосоме, а вторая задача – определить расстояние между ними. Т.е. на счет относительного расположения ген картирование более или менее справляется с этим, если посмотреть на слайд позиции этих маркеров совпадают, кроме двух они переставлены местами, очевидно здесь присутствует какая-то аномалия. Что касается расстояния между маркерами, то здесь уже все несколько хуже – вы видите что некоторые расстояния оказываются несколько больше, некоторые несколько меньше чем реальные расстояния. Соответственно сейчас все больше и больше значения приобретают методики физического картирования, которые сейчас являются неотъемлемой частью геномного проекта как при классическом подходе к секвенированию, так и при шотган подходе, потому что в большинстве случаев для ликвидации пробелов на стадии финиширования все равно без карты не обойтись, особенно для эукариотических геномов.
Популярное:
|
Последнее изменение этой страницы: 2016-07-12; Просмотров: 2427; Нарушение авторского права страницы