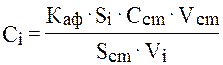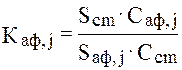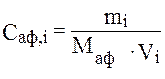|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Особенности жидкостных хроматографов
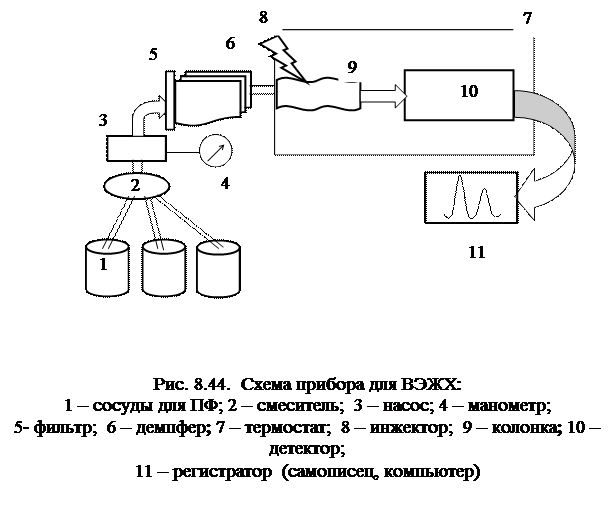
Жидкостной хроматограф (рис.8.44) - сложный прибор, поскольку система подачи элюента под давлением включает в себя ряд дополнительных узлов: устройство для приготовления элюента (обычно смеси растворителей), систему дегазации подвижной фазы, насосы и измерители давления. Насосы (создающие давление до нескольких десятков мегапаскалей) должны работать стабильно, причем скорость потока подвижной фазы (от 0, 1 до 10 мл/мин) необходимо поддерживать с большой точностью. Необходимо тщательное обезгаживание всех используемых растворителей (например, продувкой гелием). Сосуды для ПФ имеют объем 0, 7 – 1, 0 л, причем часто используют смеси растворителей, приготавливаемые в приборе по заданной программе автоматически. Их состав можно менять в процессе работы, как правило, таким образом, чтобы элюирующая сила растворителя увеличивалась. Поскольку насосы создают пульсирующие потоки, что затрудняет детектирование, для сглаживания колебаний применяют демпфирующие устройства. Инжекторы обеспечивают ввод проб размером от 0, 1 мкл до нескольких мл с высокой воспроизводимостью. Размывание пробы в инжекторе должно быть минимально.
Для непрерывного контроля состава элюата чаще всего в ВЭЖХ используют дифференциальные рефрактометры, УФ-, спектрофотометрические, люминесцентные и кондуктометрические детекторы. Единого универсального детектора для жидкостной хроматографии не существует. Дифференциальный рефрактометр – универсальный детектор, измеряющий показатель преломления системы на выходе из колонки. Его чувствительность относительно невысока (предел обнаружения около 10-6 г), а диапазон линейности составляет до 4-х порядков концентрации. Недостаток – чрезвычайная чувствительность к колебаниям температуры. УФ-детектор, настроенный на определенную длину волны, реагирует на все вещества, поглощающие при этой длине волны. Очевидно, он тем более чувствителен, чем сильнее молярные коэффициенты поглощения определяемых веществ отличаются от таковых для растворителя. Он достаточно селективен, предел обнаружения около 10-9 г, диапазон линейности до 5 порядков. Детектор используют на определенных длинах волн, чаще всего 254 нм: при этой длине волны поглощают все ароматические соединения, альдегиды, кетоны и некоторые другие вещества. Кондуктометрический детектор применяют в ионной хроматографии, поскольку его работа основана на измерении электропроводности раствора, пропорциональной числу ионов. Вольтамперометрический детектор используют для определения электроактивных веществ. Это один из самых высокочувствительных детекторов.
8.4.9. Применение хроматографии Положение пика на хроматограмме (время или объем удерживания) характеризует природу вещества и используется для качественного анализа, а площадь хроматографического пика пропорциональна количеству вещества, прошедшего через детектор. Последнее используется в количественном анализе. 8.4.9.1. Качественный анализ Идентификация определяемого вещества производится по параметрам удерживания tR или VR . Эти параметры характеризуются хорошей воспроизводимостью (sr не превышает 2 %). Как же по времени удерживания или удерживаемому объему установить природу вещества? Для этого существует несколько приемов. 1) Если круг веществ, которые могут содержаться в исследуемой пробе, не слишком широк и приблизительно известен, сравнивают параметры удерживания определяемого и “подозреваемого” веществ, полученные в одинаковых условиях. Обычно используют величины исправленных времен удерживания или удерживаемых объемов. Совпадение этих величин говорит о том, что эти вещества могут быть идентичными (но не обязательно). В хроматографии достоверен только отрицательный результат: несовпадение времен удерживания стопроцентно свидетельствует о том, что это – другое вещество. При совпадении времен удерживания желательно повторить опыт в совершенно других условиях (другая колонка, НФ, температура и т.д.). При сравнении хроматограмм, полученных на разных колонках или приборах, во избежание ошибок рекомендуется производить идентификацию по относительным параметрам удерживания, т.е. отношению удерживаемого объема определяемого компонента к удерживаемому объему вещества, принятого за стандарт: tотн = tR/ / t /R, ст. =VR// V /R, ст. Эта величина зависит только от состава подвижной и неподвижной фаз. Выбор стандарта в этом методе произволен, но его поведение должно быть хорошо известно, а параметры пика воспроизводимы. 2) Часто бывает так, что о составе анализируемого объекта вообще ничего не известно. В таком случае перебирать данные о миллионах существующих в природе веществ бессмысленно. Удобно использовать индексы Ковача (I), которые по своей сути также являются относительными параметрами удерживания. В этом случае за стандарты берут два соседних (между собой) нормальных алкана, один из которых элюируется до, а второй - после неизвестного вещества, т.е.
Например, неизвестное соединение имеет исправленное время удерживания 19, 5 мин. Исправленное время удерживания для н-гептана и н-гексана соответственно равно 29, 3 и 13, 7 мин. Используя эти данные, рассчитаем индекс Ковача по уравнению:
В справочнике находим, что полученное значение (с некоторой степенью приближения) соответствует бензолу (I = 650). Идентификация по индексам Ковача более надежна, чем по относительным параметрам удерживания, в частности потому, что они меньше подвержены влиянию температуры колонки, чем другие параметры. Однако все-таки различия температур при идентификации не должны превышать 300. Существует несколько " вспомогательных" приемов качественного анализа. 3) Неплохой способ идентификации - использование двух детекторов: универсального (катарометр, рефрактометр) и достаточно специфичного (например, электронного захвата или УФ-детектор). На хроматограмме, полученной со специфичным детектором, ряд сигналов может отсутствовать. Заранее известно, на какие классы соединений не реагирует этот детектор. Следовательно, получают дополнительную информацию о природе сигналов, зарегистрированных с помощью универсального детектора, но не проявившихся с использованием специфичного. Сравнение хроматограмм, полученных на разных детекторах, дает информацию, например, о составе органического вещества или его функциональных группах. tR/ = A + Bz Для идентификации достаточно знать параметры удерживания нескольких соединений гомологического ряда, по которым построить график такой зависимости и пользоваться им. Вместо построения графика можно рассчитать коэффициента приведенного выше уравнения (А и В). Часто приведенное время удерживания (в ГХ) линейно зависит от температуры кипения членов гомологического ряда. Этот факт также может быть использован для идентификации органических веществ. В целом следует сказать, что на практике идентификация соединений по хроматограмме не так проста, как может показаться, особенно для смесей, о составе которых заранее абсолютно ничего не известно. 8.4.9.2. Количественный анализ Количественный анализ проводят, оценивая высоту или площадь пика, поскольку они пропорциональны количеству вещества в хроматографической зоне, а следовательно, и концентрации компонента в анализируемой смеси. Наиболее совершенные приборы автоматически измеряют параметры сигналов и математически обрабатывают их. Чаще, однако, пока еще приходится делать это вручную, с помощью обычной линейки. Измерение высот пиков проще и надежнее, чем измерение площадей, особенно для веществ с малым временем удерживания (чем меньше время tR, тем острее пик). Однако площади измеряют чаще, поскольку их зависимость от концентрации, как правило, с большей степенью надежности описывается линейной функцией. Для этого используют несколько способов. 1) Для обработки симметричных пиков находят произведение высоты пика (h) на его полуширину (W'), что составляет около 84 % площади пика. Точность измерения площади пика зависит от соотношения высоты пика и его ширины (оптимальное соотношение - от 2 до 10). 2) Для несимметричных пиков проводят касательные к началу и окончанию пика и измеряют расстояние от вершины пика до этой касательной. Площадь полученного треугольника составляет около 96 % от истинной площади пика и пропорциональна количеству вещества в пробе. 3) Инструментальные методы определения площадей (с помощью планиметра или взвешиванием вырезанных по контуру пиков) применяются, главным образом, при обработке хроматограмм, имеющих асимметричные пики. Оба способа требуют аккуратности и не дают удовлетворительных результатов при малых площадях пиков. Количественный состав пробы затем рассчитывают, используя следующие методы: 1) нормировки (с использованием или без использования поправочных коэффициентов); 2) внешней стандартизации (абсолютной градуировки); 3) внутренней стандартизации. Метод нормировки. Это самый простой метод, используемый только в тех случаях, когда детектор откликается на все компоненты смеси, т.е. число сигналов равно числу компонентов. Метод нормировки без поправочных коэффициентов применим только в том случае, если детектор имеет одинаковую чувствительность к каждому из разделяемых веществ, т.е. компоненты смеси, взятые в одинаковых количествах, дают одну и ту же площадь пика (Si), что не всегда имеет место. Сущность этого метода заключается в том, что содержание (обычно - массовая доля) компонента (Рi) может быть рассчитано по формуле:
Другими словами, наличие лишь двух одинаковых сигналов на хроматограмме означает то, что в смеси присутствуют два компонента в равных количествах, три пика с отношением площадей 1: 2: 3 - три компонента с тем же соотношением их количеств и т.д. Если чувствительность детектора различна по отношению к каждому компоненту пробы (что чаще всего и бывает), то в формуле появляются поправочные коэффициенты (Кi), учитывающие чувствительность детектора к данному компоненту:
Поправочные коэффициенты получают после дополнительно проведенного хроматографического исследования эталонной смеси (известного состава) и рассчитывают по формуле:
(здесь индекс “0” означает, что смесь эталонная, индекс “ст.” относится к веществу, принятому за стандарт). Очевидно, удобно составлять стандартные смеси с равным содержанием компонентов. В таком случае
Если пики достаточно узкие и симметричные, то вместо площади пика (Si) можно использовать его высоту (hi). Величины поправочных коэффициентов зависят от типа детектора и природы разделяемых веществ. Для примера рассмотрим хроматографический анализ смеси жидких углеводородов на хроматографе ЛХМ-8МД (рис. 8.9). Анализ производился на насадочной колонке (хроматон + апиезон) длиной 2 м с помощью детектора - катарометра. По хроматограмме эталонной смеси, составленной из одинаковых количеств гексана, гептана, октана и нонана (рис.8.10), были определены поправочные коэффициенты. Результаты анализа исследуемой смеси углеводородов представлены в таблице 8.13. Поправочные коэффициенты Кi находили относительно гексана по формуле:
(здесь индекс " 0" означает, что смесь эталонная, индекс " ст" относится к веществу, принятому за стандарт). Содержание Pi (%) компонентов в анализируемой смеси определяли по формуле:
Таблица 8.13 Результаты анализа смеси углеводородов
Метод внешнего стандарта (абсолютной градуировки) - это хорошо известный метод градуировочного графика. Он предполагает раздельное хроматографирование стандартных растворов (для построения градуировочных графиков) и анализируемой смеси, поэтому условия хроматографирования должны быть предельно одинаковыми. Содержание вещества в пробе обычно определяют графически (зависимость площади пика от содержания определяемого вещества в стандартном растворе). Для построения градуировочного графика, как обычно, необходимо приготовить несколько стандартных растворов каждого из определяемых веществ. Поскольку такой прием градуировки требует много времени, его используют лишь тогда, когда требуется провести определение одного - двух компонентов, а не всего состава анализируемого объекта. В то же время следует признать, что точность получаемых результатов в данном случае обычно выше, чем точность метода нормировки. Можно несколько ускорить работу, применив вариант этого метода, напоминающий метод молярного свойства. В таком случае содержание вещества определяют по формуле: Pi (%) = где Кi - угловой коэффициент градуировочной прямой, рассчитываемый после хроматографирования стандартных растворов: Ki = Понятно, что в случае прямо пропорциональной зависимости это может быть только один стандартный раствор. Метод внутреннего стандарта . Обычно его применяют, когда некоторые компоненты смеси по той или иной причине не дают сигнала детектора, а следовательно, метод нормировки даже для определения остальных компонентов использовать нельзя. В таком случае в анализируемую смесь вводят некоторое количество постороннего стандартного вещества, не присутствующего в анализируемой пробе. Для этого выбирают соединение, которое полностью отделяется от всех имеющихся в образце компонентов и может служить стандартом. Очевидно, что оно должно быть химически инертным по отношению к компонентам смеси, его параметры удерживания - близки для таковых, а пик симметричен. Добавку выбирают такую, чтобы концентрация внутреннего стандарта была близка к концентрациям определяемых компонентов. Затем составляют различные искусственные смеси (известного состава) из стандарта и всех интересующих соединений, снимают хроматограммы таких смесей и определяют на них площади пиков. По результатам хроматографирования таких смесей рассчитывают поправочные коэффициенты (коэффициенты чувствительности) по формуле:
где Sвн.ст. и Si - площади пиков внутреннего стандарта и соответствующего определяемого компонента; Рвн.ст. и Рi - концентрации стандарта и исследуемого компонента в искусственных смесях. Если есть уверенность в строгом выполнении линейной зависимости площади пика от концентрации, достаточно одной искусственной смеси, но для повышения точности результатов поправочные коэффициенты рассчитывают по результатам хроматографирования нескольких смесей и полученные значения усредняют.
где r - отношение массы внутреннего стандарта, введенного в пробу, к массе самой пробы. Полученные данные можно обработать и графически, предварительно построив градуировочные графики для определения каждого компонента. Они строятся в координатах зависимости отношения площадей компонента и внутреннего стандарта от отношения содержаний компонента и внутреннего стандарта в искусственных смесях: Si / Sвн.ст. от Pi / Рвн.ст. Метод внутреннего стандарта, вероятно, самый трудоемкий метод (т.к. стандарт добавляется к каждому образцу, причем он должен полностью отделяться от всех компонентов пробы), но обычно и самый точный. Рассмотрим метод внутреннего стандарта на примере. Хроматографический метод часто используют в кинетических исследованиях. При этом через определенные промежутки времени отбирают необходимое количество реакционной смеси из зоны реакции для анализа. По хроматограммам анализируемых проб находят содержание (Сi) реагентов (продуктов или исходных веществ). Зависимость содержания какого-либо реагента от времени в виде кинетической кривой позволяет определить некоторые кинетические параметры изучаемой системы: скорость реакции, порядок реакции по данному реагенту, энергию активации. Эти параметры необходимы для установления механизма протекания реакции. Например, при изучении реакции между ацетофеноном (C6H5COCH3) и селенистой кислотой (H2SeO3) в присутствии хлороводородной кислоты (HCl) в среде диэтилового эфира за кинетикой процесса можно следить по изменению концентрации ацетофенона в реакционной среде во времени. К отобранным через определенные промежутки времени реакционным пробам добавляли экстрагирующий раствор толуола в эфире (толуол выполняет роль внутреннего стандарта). Хроматографический анализ эфирного слоя отмытой от следов хлороводородной кислоты пробы производился на газовом хроматографе с автоматическим дозатором. Для анализа применялась капиллярная колонка НР-5 и пламенно-ионизационный детектор. На используемом хроматографе, снабженном интегратором, определение площадей хроматографических пиков осуществлялось автоматически. В табл. 8.14 представлены результаты анализа одной из реакционных проб, соответствующей хроматограмме (рис 8.47). Содержание ацетофенона (Сi, моль/л) в реакционных пробах находили методом внутреннего стандарта по формуле:
где Si, Scm – площади пиков, соответствующих на хроматограмме ацетофенону из i-той пробы и толуолу, используемому в качестве стандарта, Сcm – содержание (моль/л) толуола в экстрагирующем растворе (толуол + диэтиловый эфир), Vcm – объем экстрагирующего раствора, который добавлен к i-той пробе; Vi – объем реакционной пробы, отобранной из реакционной зоны для анализа; Каф – чувствительность детектора хроматографа к ацетофенону относительно стандарта (толуола).
Таблица 8.14 Результаты анализа реакционной смеси
Для нахождения Каф были составлены искусственные смеси из разных количеств ацетофенона и экстрагирующей смеси, содержащей 1 % (по объему) толуола. Результаты калибровки прибора, т.е. нахождения коэффициента чувствительности пламенно-ионизационного детектора к ацетофенону относительно толуола, представлены в табл. 8.15.
Таблица 8.15 Результаты анализа искусственных смесей
Калибровочные коэффициенты Каф, j для разных искусственных смесей находили по формуле:
где Scm, Sаф, j – площади пиков толуола и ацетофенона на хроматограммах смесей; Ccm, Cаф, j – содержание (моль/л) толуола и ацетофенона в этих смесях. Молярную концентрацию ацетофенона в смесях рассчитывали по формуле: где mi – масса ацетофенона в смеси (г); Маф – молярная масса ацетофенона (моль/л); Vi – объем смеси (л). Например:
Содержание толуола в смесях можно найти по формуле:
где: Vтол = 1, 0 мл (объем толуола в экстрагирующей смеси); dтол = 0, 867 г/мл (плотность); Мтол = 92, 14 г/моль (молярная масса); Vэкс = 100 мл (объем экстрагирующей смеси). По данным табл.8.3 был найден средний коэффициент чувствительности детектора к ацетофенону относительно толуола: Каф = 0, 924. Популярное:
|
Последнее изменение этой страницы: 2016-08-24; Просмотров: 987; Нарушение авторского права страницы
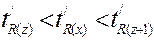 , где z - число атомов углерода в алкане. Для всех н-алканов I = 100. z, а индекс Ковача для идентифицируемого соединения может быть рассчитан по формуле:
, где z - число атомов углерода в алкане. Для всех н-алканов I = 100. z, а индекс Ковача для идентифицируемого соединения может быть рассчитан по формуле: 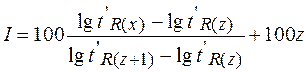
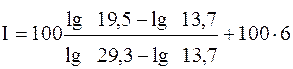 =648, 2
=648, 2

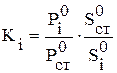
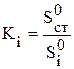
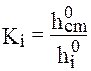
 .100%
.100% ,
, 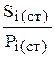
 ,
,  ,
,