|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Вопрос 4. Митохондриальные заболеванияСтр 1 из 12Следующая ⇒
Вопрос 2 Существуют следующие критерии основных типов ядерного наследования. А) Аутосомно-рецессивное наследование: 1) признак встречается относительно редко, не в каждом поколении; 2) если признак имеется у обоих родителей, то этот признак имеют все их дети; 3) признак встречается и у детей, родители которых не имеют изучаемого признака; 4) мужчины и женщины с изучаемым признаком встречаются с приблизительно одинаковой частотой. Б) Аутосомно-доминантное наследование: 1) признак встречается часто, в каждом поколении; 2) признак встречается у детей, у которых хотя бы один из родителей имеет изучаемый признак; 3) мужчины и женщины с изучаемым признаком встречаются с приблизительно одинаковой частотой. Вопрос 3. В) Сцепленное сY-хромосомой, или голандрическое, наследование: 1) признак встречается часто, в каждом поколении; 2) признак встречается только у мужчин; 3) признак передается по мужской линии: от отца к сыну и т.д. Г) Рецессивное сцепленное с Х-хромосомой наследование: 1) признак встречается относительно редко, не в каждом поколении; 2) признак встречается преимущественно у мужчин, причем у их отцов признак обычно отсутствует, но имеется у дедов (прадедов) по материнской линии; 3) у женщин признак встречается только тогда, когда он имеется и у их отца. Д) Доминантное сцепленное с Х-хромосомой наследование: 1) признак встречается часто, в каждом поколении; 2) признак встречается у детей, у которых хотя бы один из родителей имеет изучаемый признак; 3) признак встречается и у мужчин, и у женщин, но женщин с таким признаком приблизительно в два раза больше, чем мужчин; 4) если изучаемый признак имеет мужчина, то все его дочери будут иметь этот признак, а у всех его сыновей этот признак будет отсутствовать. В конкретной семье соотношение индивидуумов с изучаемым признаком и без признака может значительно отличаться от теоретически ожидаемых соотношений, характерных для определенного типа наследования. Это объясняется главным образом случайным характером распределения хромосом в процессе гаметогенеза. Тем не менее, характер родословной, особенности передачи признака (заболевания) из поколения в поколение, соответствие родословной критериям наследования того или иного типа позволяют сделать определенный вывод о типе наследования признака (патологии) в конкретной семье. Установление типа наследования заболевания часто является решающим в постановке правильного диагноза и, следовательно, в выборе эффективного лечения, проведении адекватных профилактических мероприятий и даче оправданных медико-генетических рекомендаций. Возникновение хромосомных аберраций Основной предпосылкой для возникновения хромосомных перестроек является появление в клетке двунитевых разрывов ДНК, то есть разрывов обеих нитей спирали ДНК в пределах нескольких пар оснований. Двунитевые разрывы ДНК возникают в клетке спонтанно или под действием различных мутагенных факторов: физической (ионизирующее излучение), химической или биологической (транспозоны, вирусы) природы. Двунитевые разрывы ДНК возникают запрограммированно во время профазы Iмейоза, а также при созревании Т- и B-лимфоцитов во время специфической соматической V(D)J рекомбинации. Нарушения и ошибки процесса воссоединения двунитевых разрывов ДНК приводят к появлению хромосомных перестроек. Классификация Делеции Различают терминальные (утрата концевого участка хромосомы) и интеркалярные (утрата участка на внутреннем участке хромосомы) делеции. Если после образования делеции хромосома сохранила центромеру, она аналогично другим хромосомам передается при митозе, участки же без центромеры, как правило, утрачиваются. При конъюгации гомологичных хромосом во время мейоза у нормальной хромосомы на месте, соответствующем интеркалярной делеции у дефектной хромосомы, образуется делеционная петля, которая компенсирует отсутствие делетированного участка. Врождённые делеции у человека редко захватывает протяженные участки хромосом, обычно такие аберрации приводят к гибели эмбриона на ранних этапах развития. Самым хорошо изученным заболеванием, обусловленным достаточно крупной делецией, является синдром кошачьего крика, описанный в 1963 году Жеромом Леженом. В его основе лежит делеция участка короткого плеча 5 хромосомы. Для больных характерен ряд отклонений от нормы: нарушение функций сердечно-сосудистой, пищеварительной систем, недоразвитие гортани (с характерным криком, напоминающим кошачье мяуканье), общее отставание развития, умственная отсталость, лунообразное лицо с широко расставленными глазами. Синдром встречается у 1 новорожденного из 50000. Современные методы выявления хромосомных нарушений, прежде всего флуоресцентная гибридизация in situ, позволили установить связь между микроделециями хромосом и рядом врождённых синдромов. Микроделециями, в частности, обусловлены давно описанные синдром Прадера-Вилли и синдром Вильямса. Дупликации Дупликации представляют собой класс перестроек, который объединяет как внутри-, так и межхромосомные перестройки. Вообще, любая дупликация — это появление дополнительной копии участка хромосомы, которая может располагаться сразу за тем районом, который дуплицирован, тогда это тандемная дупликация, либо в новом месте или в другой хромосоме. Новая копия может образовать отдельную маленькую хромосому со своими собственными теломерами и центромерой, тогда это свободная дупликация[3]: 2. Тандемные дупликации появляются в половых клетках при мейозе в результате неравного кроссинговера (в этом случае второй гомолог несет делецию) или в соматических клетках в результате неаллельной гомологичной рекомбинации при репарации двунитевого разрыва ДНК. В процессе кроссинговера у гетерозиготы при конъюгации хромосомы с тандемной дупликацией и нормальной хромосомы, как и при делеции, формируется компенсационная петля. Практически у всех организмов в норме наблюдается множественность генов, кодирующих рРНК (рибосомальную РНК). Это явление назвали избыточностью генов. Так у E. coli на рДНК (ДНК, кодирующее рРНК) приходится 0, 4 % всего генома, что соответствует 5-10 копиям рибосомальных генов. Другой пример дупликации — мутация Bar у Drosophila, обнаруженная в 20-х годах XX века Т. Морганом и А. Стёртевантом. Мутация обусловлена дупликацией локуса 57.0 X-хромосомы. У нормальных самок (B+/B+) глаз имеет 800 фасеток, у гетерозиготных самок (B+/B) глаз имеет 350 фасеток, у гомозигот по мутации (B/B) — всего 70 фасеток. Обнаружены также самки с трижды повторенным геном — double Bar (BD/B+). В 1970 году Сусумо Оно в монографии «Эволюция путем дупликации генов» разработал гипотезу об эволюционной роли дупликаций, поставляющих новые гены, не затрагивая при этом функций исходных генов. В пользу этой идеи говорит близость ряда генов по нуклеотидному составу, кодирующих разные продукты. Это трипсин ихимотрипсин, гемоглобин и миоглобин и ряд других белков. Инверсии Инверсией называют поворот участка хромосомы на 180°. Различают парацентрические (инвертированный фрагмент лежит по одну сторону от центромеры) и перицентрические (инвертированный фрагмент лежит по разные стороны от центромеры) инверсии. При инверсиях не происходит потери генетического материала, поэтому инверсии, как правило, не влияют на фенотип носителя. Однако, если у гетерозигот по инверсиям (то есть у организма, несущего как нормальную хромосому, так и хромосому с инверсией) в процессе гаметогенеза при мейозе происходит кроссинговер в пределах инвертированного участка, то существует вероятность формирования аномальных хромосом, что в свою очередь может привести к частичной элиминации половых клеток, а также формировании гамет с несбалансированным генетическим материалом. Более 1% человеческой популяции являются носителями перицентрической инверсии в 9 хромосоме, которую считают вариантом нормы[4]. Транслокации
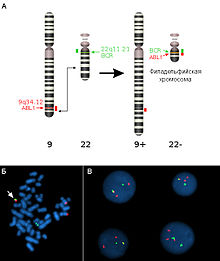
Детекция филадельфийской хромосомы при помощифлуоресцентной гибридизации in situ Транслокации представляют собой межхромосомную перестройку, при которой происходит перенос участка одной хромосомы на другую. Отдельно выделяют реципрокные транслокации (когда две негомологичные хромосомы обмениваются участками) иРобертсоновские транслокации, или центрические слияния (при этом две негомологичные акроцентрические хромосомы объединяются в одну с утратой материала коротких плеч). Первым центрические слияния описал американец У.Робертсон(W.R.B.Robertson) в 1916 г., сравнивая кариотипы близких видов саранчовых. Реципрокные транслокации не сопровождаются утратой генетического материала, их также называют сбалансированными транслокациями, они, как правило, не проявляются фенотипически. Однако, у носителей реципрокных транслокаций половина гамет несёт несбалансированный генетический материал, что приводит к снижению фертильности, повышенной вероятности спонтанных выкидышей и рождения детей с врождёнными аномалиями. Частота гетерозигот по реципрокным транслокациям оценивается как 1 на 600 супружеских пар. Реальный риск рождения детей с несбалансированным кариотипом определяется характером реципрокной транслокации (спецификой хромосом, вовлеченных в перестройку, размерами транслоцированных сегментов) и может достигать 40 %. Примером реципрокной транслокации может служить транслокация типа «филадельфийская хромосома» (Ph) между хромосомами 9 и 22. В 95 % случаев именно эта мутация в гемопоэтических клетках-предшественниках является причинойхронического миелобластного лейкоза. Эту перестройку описали П.Новелл (P.Nowell) и Д.Хангерфорд (D.Hungerford) в 1960 г. и назвали в честь города в США, где оба работали. В результате этой транслокации ген ABL1 из хромосомы 9 объединяется с геном BCR хромосомы 22. Активность нового химерного белка приводит к нечувствительности клетки к воздействию факторов роста и вызывает её безудержное деление. Робертсоновские транслокации являются одним из наиболее распространенных типов врождённых хромосомных аномалий у человека. По некоторым данным, их частота составляет 1: 1000 новорожденных. Их носители фенотипически нормальны, однако у них существует риск самопроизвольных выкидышей и рождения детей с несбалансированным кариотипом, который существенно варьирует в зависимости от хромосом, вовлеченных в слияние, а также от пола носителя. Большинство Робертсоновских транслокаций (74 %) затрагивают хромосомы 13 и 14. В структуре обращаемости на пренатальную диагностику лидерами оказываются носители der(13; 14) и der(14; 21)[5]: 1. Последний случай, а именно, Робертсоновская транслокация с участием хромосомы 21 приводит к так называемому «семейному» (наследуемому) синдрому Дауна. Робертсоновские транслокации, возможно, являются причиной различий между числом хромосом у близкородственных видов. Показано, что два плеча 2-й хромосомы человека соответствуют 12 и 13 хромосомам шимпанзе. Возможно, 2-я хромосома образовалась в результате робертсоновской транслокации двух хромосомобезьяноподобного предка человека. Таким же образом объясняют тот факт, что различные виды дрозофилы имеют от 3 до 6 хромосом. Робертсоновские транслокации привели к появлению в Европе нескольких видов-двойников (хромосомные расы) у мышей группы видов Mus musculus, которые, как правило, географически изолированы друг от друга. Набор и, как правило, экспрессия генов при робертсоновских транслокациях не изменяются, поэтому виды практически неотличимы внешне. Однако они имеют разные кариотипы, а плодовитость при межвидовых скрещиваниях резко понижена. Изохромосомы Изохромосомы состоят из двух копий одного плеча хромосомы, соединенных центромерой таким образом, что плечи образовавшейся хромосомы представляют собой зеркальные «отражения» друг друга. В определенном смысле изохромосома представляет собой гигантскую инвертированную дупликацию размером с целое плечо и делецию другого плеча. Пациенты с 46 хромосомами, из которых одна представляет собой изохромосому, являются моносомиками по генам утраченного хромосомного плеча и трисомиками по генам, присутствующим в изохромосоме. Если изохромосома является добавочной, то данный пациент является тетрасомиком по генам, представленным в изохромосоме. В целом, чем меньше изохромосома, тем меньше генетический дисбаланс, и тем более вероятно выживание плода или ребенка с такой перестройкой. Следовательно, не удивительно, что наиболее частые из описанных случаев аутосомных изохромосом вовлекают хромосомы с маленькими плечами. Некоторые из наиболее частых участников формирования изохромосом — это короткие плечи хромосом 5, 8, 12, 18[6]. Для объяснения возникновения изохромосом можно предположить два механизма: (1) вследствие аномального поперечного разделения центромеры при делении клетки или (2) в результате неправильного слияния концов изохроматидного разрыва, образовавшегося в прицентромерной области[5]: 2. Вопрос 7.
Синдром Пра́ дера — Ви́ лли — редкая генетическая аномалия. При синдроме Прадера — Вилли отсутствуют или не экспрессируются примерно 7 генов из 15-й хромосомы, унаследованной от отца. Кариотип 46 XX или ХУ, 15q-11-13. Синдром впервые описали в 1956 г. Андреа Прадер, Хайнрих Вилли, Алексис Лабхарт, Эндрю Зиглер и Гвидо Фанцони в Швейцарии. Частота встречаемости — 1: 12 000 — 15 000 живорождённых младенцев. При синдроме Прадера-Вилли страдает отцовская хромосома; в случае повреждения материнской хромосомы возникает синдром Ангельмана. Особенности Для синдрома Прадера — Вилли характерны: · до рождения: низкая подвижность плода; · часто — неправильное положение плода; · дисплазия тазобедренных суставов · ожирение; склонность к перееданию (чаще проявляется к 2-м годам); · пониженный мышечный тонус (гипотонус); пониженная координация движений; · маленькие кисти и стопы, низкий рост; · повышенная сонливость; · страбизм (косоглазие); · сколиоз (искривление позвоночника); · пониженная плотность костей; · густая слюна; плохие зубы; · сниженная функция половых желёз (гипогонадизм); в результате, как правило, бесплодие; · речевая задержка, задержка психического развития; отставание в освоении навыков общей и мелкой моторики. · более позднее половое созревание. Внешние признаки: у взрослых выражена переносица; лоб высокий и узкий; глаза, как правило, миндалевидные; губы узкие. Но все это нельзя найти в одном ребенке, чаще встречается не более пяти вышеуказанных признаков. Диагностика Синдром диагностируется путём генетического анализа, рекомендуемого для новорождённых с пониженным мышечным тонусом (гипотонусом). Иногда вместо диагноза «синдром Прадера — Вилли» врачи ошибочно ставят диагноз «синдром Дауна» (поскольку синдром Дауна встречается намного чаще). Еще часто ставят диагноз миопатия. Дети с синдромом Прадера-Вилли очень похожи между собой, опытный генетик сможет установить диагноз не дожидаясь исследования крови. Лечение Синдром Прадера — Вилли является врождённой генетической аномалией; в настоящее время специфические способы его лечения не разработаны. Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии. Рекомендуется использование специальных методик развития ребёнка, занятия с логопедом и дефектологом. Рекомендуется приём «гормонов роста», заместительная гормональная терапия (с применением гонадотропинов). Гипогонадизм обычно проявляется в микропении и неопущении яичек у мальчиков (крипторхизм); врачи могут порекомендовать подождать, пока яички опустятся сами, а также порекомендовать хирургическое вмешательство либо гормонотерапию. Для коррекции повышенного веса применяется диета с ограничением количества жиров и углеводов. Из-за ожирения, сопутствующего синдрому, нужно пристально следить за количеством и качеством пищи, поглощаемым человеком с синдромом Прадера-Вилли (обычно люди с таким синдромом способны много съесть, не наедаясь). Возможным осложнением может стать апноэ (задержка дыхания во сне). Риск Риск, что следующий ребёнок у тех же родителей родится также с синдромом Прадера — Вилли, зависит от механизма, вызвавшего генетический сбой. Этот риск меньше 1 % в случае, если у первого ребёнка делеция гена или партеногенетическая (однородительская) дисомия; до 50 % — если сбой вызван мутацией; до 25 % — в случае транслокации родительских хромосом. Родителям рекомендуется пройти генетическое обследование. Синдром Ангельмана — генетическая аномалия. Для него характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки. При синдроме Ангельмана отсутствуют некоторые гены из 15-й хромосомы (в большинстве случаев — частичная делеция либо другая мутация 15 хромосомы). При синдроме Ангельмана страдает материнская хромосома; в случае повреждения отцовской хромосомы возникает синдром Прадера-Вилли. Кариотип 46 XX или XY, 15р−. Обычно синдром вызывается спонтанным хромосомным дефектом, когда отсутствует большая смежная область из 3—4 миллионов пар оснований ДНК в области q11—q13 15-й хромосомы. Согласно результатам многих независимых исследований, причиной возникновения синдрома Ангельмана может являться мутацияв гене UBE3A. Продукт этого гена — ферментный компонент сложной системы деградации белков [1] Синдром назван по имени британского педиатра Гарри Ангельмана, впервые описавшего его в 1965 г. Частота встречаемости, по разным данным, — 1: 10 000—20 000 живорожденных младенцев.[2] Однако, согласно данным Центра развития человека и отклонений в развитии (университет Вашингтона, США), можно предполагать, что доля людей с синдромом Ангельмана в действительности намного больше статистической.[источник не указан 1448 дней] Особенности Для синдрома Ангельмана характерны: · В 75 % проблемы с питанием, особенно с грудным вскармливанием, такие младенцы плохо набирают вес; · задержка в развитии навыков общей моторики (умение сидеть, ходить); · задержка речевого развития, неразвитая речь (у всех детей); · дети больше понимают, чем могут сказать или выразить; · дефицит внимания и гиперактивность; · сложности с обучением; · эпилепсия (80 % случаев), нарушения выявляются также при электроэнцефалографии; считается, что у детей с синдромом Ангельмана наблюдается вторичная (симптоматическая) эпилепсия; · необычные движения (мелкий тремор, хаотические движения конечностей); · частый смех без повода; · ходьба на негнущихся ногах — из-за этой особенности детей с этим синдромом иногда сравнивали с марионетками; · размер головы меньше среднего, нередко с уплощением затылка; · иногда характерные черты лица — широкий рот, редко расположенные зубы, выдающийся вперед подбородок, высунутый наружу язык; · нарушения сна; · страбизм (косоглазие) в 40 % случаев; · сколиоз (искривление позвоночника) в 10 % случаев; · повышенная чувствительность к высокой температуре; · чувствуют себя комфортнее в воде. Диагностика Синдром диагностируется путем генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи. Родители и врачи должны обратить внимание на случаи мелкого тремора, хаотические, порывистые движения конечностей, походку с негнущимися ногами; в ряде случаев специфическое выражение лица, слишком частый смех. Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15q11—q13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE3A. Существует небольшая группа людей, у которых результаты всех вышеописанных анализов в норме, однако наблюдаются все внешние проявления синдрома Ангельмана. Так же эту болезнь называют «синдром Петрушки» или синдромом «смеющейся куклы». Лечение Синдром Ангельмана является врожденной генетической аномалией; в настоящее время специфические способы его лечения не разработаны. Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии). Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0.3 мг мелатонина за 30 минут-1 час перед сном улучшает сон пациентов с синдромом Ангельмана. Нарушения стула регулируются назначением легких слабительных. Приступы лечатся так же, как эпилепсия. Дети с синдромом Ангельмана часто испытывают больше одного типа приступов. Показана электроэнцефалография. Нежелательное поведение. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложность в общении с другими людьми. Врачи США показывают, что для аутичных детей внутривенные инъекции гормона секретин (найденного в поджелудочной железе) успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана.
Риски Оценка риска повторного рождения ребенка с синдромом Ангельмана у тех же родителей очень сложна, необходима консультация профессионального генетика. Считается, что обычная делеция является спонтанной, риск повтора меньше 1 %. В случае молекулярной микроделеции в 15q11-q13, если она наблюдается и у матери, риск теоретически до 50 %. Мутации внутри гена UBE3A могут быть случайными и неунаследованными, в этой ситуации риск повтора < 1 %; однако эти мутации можно унаследовать от нормальной матери, и тогда теоретический риск 50 %. Партеногенетическая дисомия 15-ой пары — случайная ситуация; риск повтора < 1 %. Есть несколько людей с AS с необычными преобразованиями хромосомы 15, включая область 15q11-q13; в этих случаях оценка риска повтора зависит от хромосомных нарушений у родителей. Перспективы развития Дети с синдромом Ангельмана понимают намного больше, чем могут сказать. В некоторых случаях у них вообще нет речи; описаны дети со словарным запасом около 5-10 слов. При этом дети/люди с синдромом Ангельмана любят общаться с людьми, играть, как правило, они дружелюбны и милы. Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам [3], направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты. Перспективы развития зависят от степени пораженности хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае стала мутация), некоторые никогда не смогут ходить и говорить (это обычно происходит в случае делеции части хромосомы). С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана в период полового созревания могут участиться припадки. Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, некоторые — и ночью. Некоторые люди с синдромом Ангельмана способны есть при помощи ножа и вилки, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний». Во взрослом возрасте может появиться ожирение и ухудшиться ситуация со сколиозом. Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки. Описан один случай беременности женщины с синдромом Ангельмана: она родила девочку с таким же диагнозом. Вопрос 8. Синдро́ м Да́ уна (трисомия по хромосоме 21) — одна из форм геномной патологии, при которой чаще всего кариотип представлен 47хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями (трисомия, см. также плоидность). Существует ещё две формы данного синдрома: транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Y-хромосому) — 4 % случаев, и мозаичный вариант синдрома — 5 %. Синдром получил название в честь английского врача Джона Дауна, впервые описавшего его в 1866 году. Связь между происхождением врождённого синдрома и изменением количества хромосом была выявлена только в 1959 году французским генетиком Жеромом Леженом. Слово «синдром» означает набор признаков или характерных черт. При употреблении этого термина предпочтительнее форма «синдром Дауна», а не «болезнь Дауна»[1]. Первый Международный день человека с синдромом Дауна был проведён 21 марта 2006 года. День и месяц были выбраны в соответствии с номером пары и количеством хромосом. Патофизиология
Кариотип (хромосомный набор) человека с синдромом Дауна. В 21-й паре три хромосомы вместо двух Синдром Дауна — хромосомная патология, характеризующаяся наличием дополнительных копий генетического материала 21-й хромосомы, либо целой хромосомы (трисомия), либо её участков (например, за счёт транслокации). Последствия от наличия дополнительной копии сильно различаются в зависимости от количества дополнительного генетического материала, генетического окружения и чистой случайности. Синдром Дауна встречается как у людей, так и у других видов (например, был обнаружен у обезьян и мышей). В 2005 году британские исследователи получили анеуплоидных трансгенных мышей с наличием 21-й человеческой хромосомы в дополнение к стандартному набору мышей[6]. Нормальный человеческий кариотип содержит 46 хромосом и обозначается 46, XY у мужчин и 46, XX у женщин, в то время как у больных синдромом Дауна с трисомией по 21-ой хромосоме кариотип содержит 47 хромосом. Трисомия Трисомия — это наличие трёх гомологичных хромосом вместо пары в норме. Риск рождения ребёнка с синдром Дауна и другими численными хромосомными аномалиями растёт с возрастом матери. Точная причина этого неизвестна, но, по-видимому, она связана с возрастом яйцеклеток матери. Трисомия происходит из-за нерасхождения хромосом во время мейоза, в результате чего возникает гамета с 24 хромосомами. При слиянии с нормальной гаметой противоположного пола образуется зигота с 47 хромосомами, а не 46-ю, как без трисомии. Трисомия 21-й хромосомы в 95 % случаев является причиной возникновения синдрома Дауна, и в 88 % случаев из-за нерасхождения материнских гамет и в 8 % — мужских. Мозаицизм Трисомия обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (гамет), в этом случае все клетки организма ребёнка будут нести аномалию. При мозаицизме же нерасхождение возникает в клетке зародыша на ранних стадиях его развития, в результате чего нарушение кариотипа затрагивает только некоторые ткани и органы. Данный вариант развития синдрома Дауна называется «мозаичный синдром Дауна» (46, XX/47, XX, 21). Данная форма синдрома является, как правило, более лёгкой (в зависимости от обширности изменённых тканей и их расположения в организме), однако более трудна для пренатальной диагностики. По данному типу синдром появляется в 1—2 % случаев. Формы синдрома Дауна Примерно в 91 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребёнка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребёнка[7]. Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет[8]. Диагностика
Клинодактилия Беременная женщина может пройти обследование на выявление нарушений плода. Многие стандартные дородовые обследования способны обнаружить синдром Дауна у плода. Например, имеются специфические УЗИ-признаки синдрома. Генетические консультации с генетическими тестами (амниоцентез, биопсия хориона, кордоцентез), как правило, предлагаются семьям, риск рождения в которых ребёнка с синдромом Дауна наиболее велик. В США инвазивные и неинвазивные обследования доступны для всех женщин, вне зависимости от их возраста. Однако инвазивные обследования проводить не рекомендуется, если женщине больше 34 лет и неинвазивные обследования не показали вероятных нарушений.
Ребёнок с характерными чертами, присущими синдрому Дауна (эпикантус, плоское лицо, открытый рот, увеличенный язык, маленький нос и т. д.) Амниоцентез и биопсия хориона считаются инвазивными обследованиями, так как при них в матку женщины вводят различные инструменты, что несёт в себе некоторый риск повреждения стенки матки, плода или даже выкидыша. Риск выкидыша при биопсии хориона — 1 %, при амниоцентезе — 0, 5 %. Существует несколько неинвазивных обследований, они, как правило, проводятся в конце первого или в начале второго триместра. В каждом из них есть шанс получить ложноположительный результат, то есть обследование покажет, что у плода синдром Дауна, хотя на самом деле он здоров. Даже с самыми лучшими обследованиями вероятность обнаружения синдрома с помощью этих обследований составляет 90—95 %, а уровень ложноположительных результатов 2—5 %. Несколько неинвазивных пренатальных тестов, которые включают секвенирование фрагментов ДНК плода в материнской крови, были разработаны. Их выпускают компании Verinata, поглощенная компанией Illumina в январе 2013 года, Ariosa Diagnostics иNatera.[9] Тест MaterniT21 PLUS, представленный компанией Sequenom в октябре 2011, [9] способен детектировать синдром Дауна на основе ДНК плода в материнской крови в 209 из 212 случаев (98.6 %).[10] Международное общество пренатальной диагностики полагает, что этот современный тест может быть использован, вместе с генетическим консультированием, в случаях, когда существующие стратегии скрининга показывают у плода высокий риск развития синдрома. Хотя тест эффективен в диагностике синдрома Дауна, он не определяет другие болезни и состояния, которыем могут детектировать инвазивные тесты.[11] На данный момент амниоцентез считается самым точным обследованием. Для получения результатов у женщины требуется взять на анализ амниотическую жидкость, в которой позже выявляют клетки плода. Лабораторные работы могут занять несколько недель, но вероятность правильного результата — 99, 8 %. Ложноположительный показатель очень низок.
Прогноз Высокие риски заболеваемости обуславливают то, что средняя продолжительность жизни людей с синдромом Дауна несколько короче продолжительности жизни людей со стандартным набором хромосом. Одно исследование[источник не указан 681 день], проведённое в США в 2002 году, показало, что средняя продолжительность жизни больных — 49 лет. Тем не менее, нынешняя продолжительность жизни значительно возросла по сравнению с 25 годами в 1980-х годах. Причины смерти также изменились со временем, хронические нейродегенеративные заболевания становятся всё более распространены по мере старения населения. Большинство людей с синдромом Дауна, которые доживают до возраста 40-50 лет, начинают страдать от болезни Альцгеймера — деменции. Вопрос 9. Синдром Пата́ у (трисомия 13) — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13. История Трисомия 13 впервые описана Эразмусом Бартолином в 1657. Хромосомную природу заболевания выявил доктор Клаус Патау в 1960. Заболевание названо в его честь. Синдром Патау также был описан для племен с островов Тихого океана. Считается, что эти случаи были вызваны радиационным заражением, появившимся в результате испытаний ядерного оружия в регионе. Генезис Встречается с частотой 1: 7000-1: 14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия иробертсоновская транслокация. Другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслокационных не различается. 75 % случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае синдрома Дауна. 25 % случаев СП — следствие транслокации с вовлечением хромосом 13-й пары, в том числе в трех из четырёх таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13-й пары имеет наследственный характер с возвратным риском 14 %. Соотношение полов при синдроме Патау близко к 1: 1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25 — 30 % ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38, 5 недель). Проявления заболевания Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау. При синдроме Патау наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются Популярное:
|
Последнее изменение этой страницы: 2016-04-11; Просмотров: 1015; Нарушение авторского права страницы