
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Существует функция состояния термодинамической системы – энтропия. При обратимых процессах в изолированной системе ее энтропия не изменяется, а при необратимых – увеличивается.
В ходе самопроизвольного процесса энтропия изолированной системы должна возрастать, достигая максимального значения при равновесии. Выясним количественную меру энтропии:
или в интегральном виде:
Выражения (II, 1) и (II, 1а) являются определениями функции S, которая называется энтропией. Энтропия является однозначной, непрерывной и конечной функцией состояния. Энтропия измеряется в тех же единицах, что и теплоемкость, т. е. в джоулях на моль на кельвин ( Как указывалось выше, элементарная теплота не является в общем случае дифференциалом функции. Из уравнения (II, 1) следует, что dQ после деления на Т становится дифференциалом функции, т. е. с математической точки зрения, 1/Т является для теплоты интегрирующим множителем (или Т – интегрирующим делителем). Исследуя энтропию, очевидно, можно предсказывать направление процесса. Если в изолированной системе для какого-либо процесса энтропия возрастает, то процесс возможен (может протекать самопроизвольно); если энтропия изолированной системы согласно расчету должна убывать, то процесс невозможен (отрицателен). При постоянстве энтропии – процесс равновесен, система бесконечно близка к равновесию. Обобщая сказанное, можно записать следующее выражение:
Следует подчеркнуть, что в системе, обменивающейся теплотой и работой с окружающей средой, возможны процессы, сопровождающиеся как возрастанием, так и убылью энтропии системы. Поэтому для однозначного решения вопроса о направлении процесса следует включить в систему все тела, участвующие в процессе, и таким образом сделать систему изолированной. Методы расчета энтропии Уравнения (II, 1) и (II, 1а), определяющие энтропию, являются единственными исходными уравнениями для термодинамического расчета изменения энтропии системы. Заменяя элементарную теплоту в уравнении (II, 1а) ее выражениями через калорические коэффициенты (см. уравнения (I, 10) и (I, 10а)), получаем для равновесных процессов: dS = dS = Уравнения (II, 3) и (II, 3а) являются полными дифференциалами энтропии как функции переменных V, Т или P, Т. Коэффициенты этих уравнений – частные производные энтропии по соответствующим переменным. Подставив в уравнение (II, 3) значения калорических коэффициентов для одного моля идеального газа: l = P = RT/V [уравнение (I, 24)] и h = –V = –RT/P [уравнение (I, 25)] и полагая СV и СP независимыми от температуры (что допустимо лишь в небольших температурных интервалах), получим после интегрирования в известных пределах: DS = DS = – Применим полученные выше соотношения для расчетов изменения энтропии при некоторых процессах: 1. Фазовые превращения (изотермические процессы; Т = const): S2 – S1 = DS = Так, например, теплота плавления бензола равна 9, 764 кДж/моль; температура плавления tпл. = 5, 5°С (Т = 278, 5 К). Следовательно, изменение энтропии 1 моль бензола при плавлении (энтропия плавления) равно: DSпл. = 2. Нагревание при постоянном давлении (изобарный процесс; P = const). Из уравнений (I, 18а) и (II, 1а)получаем: DS = Найдем изменение энтропии одного моля алюминия при нагревании от 25 до 600°С. Истинная мольная теплоемкость алюминия может быть выражена уравнением: Ср= 565, 5 + 0, 290 Т. По уравнению (II, 6) изменение энтропии будет равно: DS = Постулат Планка. Абсолютные значения энтропии По уравнению (II, 3) невозможно вычислить абсолютное значение энтропии системы. Такую возможность дает новое, недоказуемое положение, не вытекающее из двух законов термодинамики, которое было сформулировано М.Планком (1912). Согласно этому положению, называемому постулатом Планка, энтропия индивидуального кристаллического вещества при абсолютном нуле равна нулю: S0 = 0 Строго говоря, постулат Планка справедлив только для индивидуальных веществ, кристаллы которых идеально построены (в кристаллической решетке все узлы заняты молекулами или атомами, правильно чередующимися и закономерно ориентированными). Такие кристаллы называются идеальными твердыми телами. Реальные кристаллы не являются таковыми, так как их кристаллическая решетка построена не идеально. Энтропия кристаллической решетки, построенной в некоторой степени беспорядочно, больше энтропии идеально построенной кристаллической решётки. Поэтому реальные кристаллы и при 0 К обладают энтропией, большей нуля. Однако энтропии реальных хорошо образованных кристаллов индивидуальных веществ при абсолютном нуле невелики. В соответствии с постулатом Планка уравнение (II, 6) для идеального твёрдого тела примет вид:
Постулат Планка используется при термодинамическом исследовании химических процессов для вычисления абсолютных значений энтропии химических соединений – величин, которые имеют большое значение при расчете химических равновесий. Энтропия широко используется в технической термодинамике (теплотехнике), как один из важных параметров рабочего тела в тепловой машине, например, водяного пара. Величины энтропии водяного пара в данном состоянии вычисляются по сравнению с некоторым стандартным состоянием – обычно 0°С и 1 amм. Эти значения энтропии используются для построения так называемых энтропийных диаграмм состояния водяного пара в координатах S-Т или S-H (диаграмма Молье). В таких диаграммах подобно диаграммам V-P можно изображать различные процессы, протекающие в рабочем теле тепловой машины и составляющие рабочие циклы машины. В заключение следует отметить, что нам не придется углубляться в область термодинамики. Наша цель лишь проиллюстрировать основные идеи этой науки и объяснить причины, по которым возможно основываться на её аргументах. Наконец, два закона термодинамики часто формулируют так: Первый закон: Энергия Вселенной всегда постоянна. Второй закон: Энтропия Вселенной всегда возрастает. Несмотря на некоторые недостатки приведенных выше формулировок обоих законов, они легко запоминаются и дают представление, о чем собственно идёт речь. Напомним, что второй закон термодинамики определяет критерии самопроизвольного протекания процессов в изолированных системах. Однако подобные условия (отсутствие обмена энергией и веществом с окружающей средой) реализуются сравнительно редко. Поэтому представляется важным сформулировать подобного рода критерии для закрытых систем, где возможен обмен энергией с окружающей средой. Для этого нам потребуется определить две новые функции состояния – энергию Гельмгольца и энергию Гиббса.
ГЛАВА III. Энергия Гельмгольца Работа процесса в общем случае, как это уже говорилось, зависит от пути процесса. Работа неравновесного процесса меньше, чем работа равновесного процесса, протекающего между теми же начальным и конечным состояниями системы. В самом деле, исходя из уравнения первого закона термодинамики (I, 2) и уравнения (II, 2), получаем в общем случае: δ W = dQ – dU £ TdS – dU (III, 1) Величина правой части этого уравнения не зависит от того, равновесен или неравновесен процесс. В случае равновесного процесса: dW = dWравн. = TdS – dU (III, 1а) Для неравновесного процесса: dW < TdS – dU (III, 1б) Сравнивая уравнения (III, 1а) и (III, 1б), получаем: dWравн. > dW Таким образом, работа равновесного процесса максимальна. Максимальная работа не зависит от пути, а определяется лишь начальным и конечным состояниями системы. Так, при S = const (равновесный адиабатный процесс) dW = –dU и Wмакс. = U1 – U2 т. е. величина максимальной работы определяется изменением внутренней энергии системы. Интегрируя при постоянной Т уравнение (III, 1а), получаем: Wмакс. = T (S2 – S1) – (U2 – U1) (III, 2) или Wмакс. = (U1 – TS1) – (U2 – TS2) (III, 2а) Выражения, стоящие в скобках, являются функциями состояния системы. Введя в уравнение (III, 2а) обозначение F º U – TS (III, 3) получаем (при T = const) Wмакс. = F1 – F2 = –DF (III, 4) где F – функция состояния, называемая энергией Гельмгольца. Таким образом, максимальная работа при изохорно-изотермических равновесных процессах равна убыли энергии Гельмгольца системы. Переписав уравнение (III, 3) в виде U = F + TS можно рассматривать внутреннюю энергию, как состоящую из двух частей – свободной энергии F и связанной энергии TS. Лишь часть внутренней энергии – свободная энергия, которую система отдает вовне при T = const, может превратиться в работу (условием для такого превращения является равновесность процесса; в неравновесном процессе свободная энергия частично или полностью переходит в теплоту). Другая часть внутренней энергии – связанная энергия – при изменении системы при Т = const не дает работы, а переходит только в теплоту: Энтропия есть, таким образом, фактор ёмкости связанной энергии. Для процессов, протекающих с изменением температуры (T Полный дифференциал функции F можно получить, дифференцируя уравнение (III, 3): dF º dU – TdS – SdT (III, 5) Сопоставив это уравнение с уравнениями (III, 1а) и (III, 1б), получим в общем виде: dF £ -SdT – dW (III, 5а) Откуда при Т = const (dF)T £ –dW (III, 6) или F2 – Fl = DF < -W; Fl – F2 > W (III, 6a) Выражение (III, 6a) отражает уже известное нам положение, что работа неравновесного процесса меньше работы равновесного процесса. Если при равновесном процессе совершается только работа расширения (dW = PdV), то из уравнения (III, 5а) получаем: dF = -SdT – PdV (III, 7) Это выражение является полным дифференциалом функции F при переменных V и Т. Полагая T = const и V = const, а также при условии отсутствия всех видов работы (dW = 0), получаем из уравнения (III, 5а): ( т. е., энергия Гельмгольца системы, находящейся при постоянных V и Т не изменяется при равновесных процессах, при неравновесных процессах ее значение убывает. Так как система, в которой протекают (и могут протекать) только равновесные процессы, бесконечно близка к равновесию, то сформулированные свойства энергии Гельмгольца позволяют судить о том, находится ли данная система в равновесии или нет. В последнем случае направление неравновесного процесса определяется убылью энергии Гельмгольца при постоянных температуре и объеме системы. Условия, которым должны удовлетворять процессы, для того чтобы по изменениям величины F можно было судить о направлении этих процессов, иные, чем для энтропии. Для энтропии это были условия постоянства внутренней энергии и объема (изолированная система), для энергии Гельмгольца это условие постоянства объёма и температуры – легко измеримых параметров системы. Энергия Гельмгольца, являясь производным понятием по отношению к энтропии, представляет собой практически более удобный критерий направления процессов, чем энтропия. Изложенные соображения могут быть выражены следующим положением: энергия Гельмгольца системы, находящейся при постоянных объёме и температуре, стремится уменьшиться при неравновесных (самопроизвольных) процессах. Когда она достигает минимального значения, совместимого с данными V и Т, система приходит в состояние равновесия. Энергия Гиббса Желая учесть в общей форме другие виды работы, кроме работы расширения, представим элементарную работу как сумму работы расширения и других видов работы: где dW' – сумма элементарных работ всех видов, кроме работы расширения. Мы назовем эту величину элементарной полезной работой, а величину W' – полезной работой. Из уравнений (III, 8) и (III, 1) получаем: dW' £ TdS – dU – PdV (III, 9) Отсюда можно найти величину W', получаемую при переходе системы из состояния 1 в состояние 2, интегрируя это уравнение в соответствующих пределах при постоянных температуре и давлении: Сгруппировав все величины, относящиеся к одному состоянию, получим: W' £ (U1 – TS1+ PV1) – (U2 – TS2 + PV2)(III, 10) Обозначим через G выражения, стоящие в скобках правой части уравнения, которые являются функциями состояния, т. е. G º U – TS + PV º H – TS (III, 11) Тогда уравнение (III, 10) можно записать следующим образом: W' £ G1 – G2 = -DG (III, 10а) Так как DG не зависит от пути процесса, то, при условии постоянства P и Т, для равновесных процессов W' будет максимально: W'макс. = G1 – G2 = –DG (III, 12) где G – функция состояния, определяемая равенством (III, 11) и называемая энергией Гиббса. Таким образом, максимальная полезная работа при изобарно-изотермических процессах равна убыли энергии Гиббса. Для получения полного дифференциала функции G при переменных P и Т дифференцируем уравнение (III, 11): dG = dU – Т dS – SdT + PdV + VdP Так как dU £ TdS – PdV – dW', то dG £ -SdT + VdP – dW' (III, 13) Из этого уравнения при постоянных Т и P получаем уравнение (III, 10а) в дифференциальной форме. При отсутствии всех видов работы, кроме работы расширения (dW' = 0), в общем случае: dG £ -SdT + VdP (III, 13а) а для равновесных процессов dG = -SdT + VdP (III, 13б) Энергия Гиббса системы при постоянных P и Т уменьшается при неравновесных (самопроизвольных) процессах, при равновесии её значение остаётся постоянным. Очевидно, равновесное состояние системы при данных P и Т соответствует минимуму энергии Гиббса. Популярное:
|
Последнее изменение этой страницы: 2016-05-29; Просмотров: 1134; Нарушение авторского права страницы

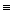 dS (II, 1)
dS (II, 1)
 ).
). (II, 2)
(II, 2)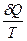 =
= 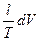 +
+ 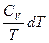 (II, 3)
(II, 3)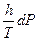 +
+ 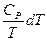 (II, 3а)
(II, 3а)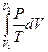 +
+ 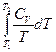 =
= 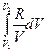 +
+  R ln
R ln 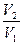 +
+ 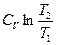
 (II, 4)
(II, 4)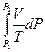 +
+ 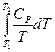 = -
= - 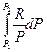 +
+ 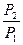 + CP ln
+ CP ln 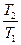 (II, 4а)
(II, 4а)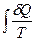 =
= 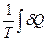 =
= 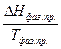 (II, 5)
(II, 5)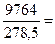 35, 06 Дж/моль
35, 06 Дж/моль 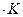
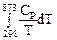 = 565, 5
= 565, 5 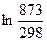 + 0, 290(873 – 298) = 607, 8 + 166, 8 = 774, 6 Дж/мольK
+ 0, 290(873 – 298) = 607, 8 + 166, 8 = 774, 6 Дж/мольK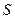 =
= 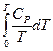 (II, 7)
(II, 7) const), деление внутренней энергии на свободную и связанную не может быть проведено и, следовательно, сами термины не имеют общего значения. Поэтому будем пользоваться для функции F названием энергия Гельмгольца.
const), деление внутренней энергии на свободную и связанную не может быть проведено и, следовательно, сами термины не имеют общего значения. Поэтому будем пользоваться для функции F названием энергия Гельмгольца. F)V, T £ 0
F)V, T £ 0