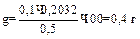|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
Определение граммового содержания меди в растворе сульфата меди (определение окислителей).
Определение меди основано на реакции: 1) 2 CuSO4+4KI↔ 2CuI+I2+2K2SO4 2│ Cu2++ẽ → Cu+ │ 2I—2 ẽ → I20 1 I2+2Na2S2O3→ 2NaI+Na2S4O8 I2+2ẽ → 2I- 2S2O32—2ẽ → S4O82- I2+2S2O32→ 2I-+ S4O82- Из первого уравнения видно, что ион Сu2+ восстанавливается до Сu+ с образованием осадка иодистой меди. Эквивалент меди в этой реакции равен его атомной массе. М(э) Сu=Аr=63, 54г, М(э) CuSO4•5H2O=249, 7г При этом йод выделяется в свободном виде. Количество выделяющегося йода эквивалентно количеству окислителя (двухвалентной меди). Выделившийся йод оттитровывают тиосульфатом натрия. Точку эквивалентности определяют с помощью индикатора крахмала. Равновесие в обратимой реакции (1) сдвинуто вправо вследствие образования осадка CuJ. Чтобы реакция практически протекала до конца, необходимо прибавить большой избыток KJ. Небольшая кислотность раствора ускоряет реакцию, подавляет гидролиз солей Cu2+. Выполнение определения 1 Анализируемый раствор доводят в мерной колбе на 100 мл до метки дистиллированной водой. 2 В колбу для титрования наливают 10 мл 10% раствора KJ, 3 мл 2н раствора H2SO4, прибавляют аликвотную часть раствора соли меди (пипетку 10 мл). Смесь оставляют на 5 минут в темноте. 1. Выделившийся свободный йод титруют раствором тиосульфата натрия. Титрование проводят точно также, как и при установлении нормальной концентрации тиосульфата натрия. Образовавшийся бледно-желтый осадок CuJ не мешает титрованию. Титрование проводят три раза, результат заносят в журнал:
V CuSO4, мл V Na2S2O3, мл индикатор 10, 0 1 _________ крахмал 10, 0 2__________ 10, 0 3__________ __________________ Vсреднее
Вычисление граммового содержания Cu2+ в растворах 1 Способ:
2 Способ:
Вопросы к разделу
1. Какой процесс лежит в основе методов окисления-восстановления? 2. Почему в ряде окислительно-восстановительных методов недопустима замена одного окислителя (восстановителя) другим окислителем (восстановителем)? 3. Какие реакции лежат в основе методов: 4. Какая из количественных характеристик раствора меняется в процессе редоксиметрического титрования? 5. Как меняется в процессе редоксиметрического титрования потенциал пары, выступающей в роли: 6. какие значения приобретают потенциалы взаимодействующих окислительно-восстановительных пар в точке эквивалентности? 7. Что представляют собой редоксиндикаторы? С чем связанно изменение их окраски? 8. Что такое область перехода окраски редоксиндикатора? Ее математическое выражение? 9. В чем заключаются достоинства и недостатки редоксиндикаторов? 10. Что представляют собой специальные индикаторы, используемые в редоксиметрическом титровании? 11. С какой целью прибегают к построению кривых титрования в редоксиметрии? 12. Почему до момента эквивалентности потенциала раствора вычисляют по концентрациям окислительно-восстановительной пары титруемого раствора, а после момента эквивалентности по концентрациям окислительно-восстановительной пары титранта? 13. Что такое «скачок» на кривой титрования? Какому процентному содержанию взаимодействующих компонентов соответствуют точки начала и конца скачка? 14. Каким образом величина скачка на кривой редоксиметрического титрования зависит от стандартных потенциалов реагирующих окислительно-восстановительных пар? 15. В чем заключается отличительная особенность кривой редоксиметрического титрования по сравнению с кривой кислотно-основного титрования? 16. Каким образом может быть увеличен скачок на кривой редоксиметрического титрования? С какой целью прибегают к его увеличению? 17. Как определяется грамм-эквивалент в редоксиметрии? 18. Какие из веществ в качестве стандартных могут быть использованы для установления нормальности раствора перманганата калия? 19. Какие условия необходимо создать для количественного определения железа (II) в растворе его соли при титровании раствором перманганата калия? 20. Чем объясняется возможность двоякого использования окислительно-восстановительной пары I2/2I- в титриметрическом анализе? 21. Какие условия необходимы для возможно более полного протекания реакции 22. Почему вдруг при титровании иода восстановителем индикатор (крахмал) вносят в титруемый раствор в конце титрования? 23. Почему иодометрические определения нельзя проводить в сильно щелочной среде? 24. Почему иодометрические определения нельзя проводить при нагревании? 25. В каких условиях проводится иодометрическое определение окислителей? Почему в данном случае необходимо использовать метод замещения? 26. Какие условия необходимы для иодометрического определения восстановителей? 27. Почему нельзя готовить по точной навеске титрованные растворы: а)перманганата калия; б) тиосульфата натрия? 28. Какие из веществ в качестве стандартных могут быть использованы для установления нормальности раствора тиосульфата натрия? 29. Почему невозможно прямое титрование раствора бихромата калия тиосульфатом натрия? 30. Почему установление нормальности раствора тиосульфата натрия по бихромату калия необходимо проводить в присутствии избытка иодистого калия? 31. Почему установление нормальности раствора тиосульфата натрия по бихромату калия недопустимым является как недостаточное количество кислоты, так и избыток ее?
32. Почему при иодометрическом определении окислителей исследуемый раствор перед титрованием необходимо выдержать в течении нескольких минут в закрытом пробкой сосуде, помещенном в темное место? 33. В чем заключается причина расхождения между предполагаемым и действительным направлением реакции иодометрического определения меди? 34. Почему при иодометрическом определении меди необходимо прибавлять к исследуемому раствору большое количество иодистого калия? 35. Почему иодометрическое определение меди необходимо проводить в слабокислой среде? Как мешают этому определению: 36. С какой целью перед ферролметрическим определением ванадия в исследуемый раствор добавляют смесь серной и фосфорной кислот? 37. Почему при феррометрическом определении хрома необходимо подкислять исследуемый раствор?
МЕТОДЫ ТИТРИМЕТРИЧЕСКОГО АНАЛИЗА, ОСНОВАННЫЕ НА РЕАКЦИЯХ КОМПЛЕКСООБРАЗОВАНИЯ.
Комплексометрическое титрование основано на реакциях, при которых определяемые ионы образуют комплексные соединения. Если получаются внутрикомплексные соединения, хелаты (с комплексонами), то применяют хелатометрическое титрование. Комплексон- это группа органических соединений-производных аминополикарбоновых кислот. Наиболее широко применяют двухнатриевую соль этилендиаминтетрауксусной кислоты-комплексон-III(ЭДТА, трилон Б).
Комплексон III содержит наряду с карбоксильными группами-СООН и третичные аминогруппы. Комплексон III образует с катионом прочные, растворимые в воде внутрикомплексные соли. При этом катион металла замещает атомы водорода карбоксильных групп-СООН и одновременно взаимодействует с азотом посредством координационной связи.
Индикаторы комплекснометрического титрования В качестве металлиндикатора используют в основном хромоген черный (эрихром черный Т) (C20H13O7N3S) и мурексид. Хромоген черный. Анион этого красителя HInd2- имеет в щелочной среде синюю окраску. С катионами двухвалентных металлов (Mg2+, Ca2+, Zn2+, Cd2+, Co2+, Ni2+) он образует комплексы винно-красного цвета: Me2++HInd2- → MeInd- +H+ синий винно-красный При последующем титровании комплексоном III эти комплексы разрушаются. Ионы металла связываются комплексоном в более прочные комплексные соединения, а анионы индикатора переходят в раствор, сообщая ему синюю окраску: MeInd-+[H2Y]2-→ [MeY]2-+ HInd2-+H+ Винно-красный бесцветный бесцветный синий В результате этого винно-красная окраска раствора сменяется синей, рН=8-10. Поэтому к титрованному раствору добавляют аммонийную буферную смесь (NH4OH+NH4Cl), которая нейтрализует выделяющиеся при этом ионы водорода. Мурексид – аммонийная соль пурпурной кислоты, комплексы металлов с мурексидом окрашены в красный цвет, менее устойчивы, чем комплексы металлов с комплексоионом III. При титровании катионы связываются комплексоионом, а анионы индикаторы освобождаются и сообщают раствору сине-фиолетовую окраску. MeInd++[H2Y]2-→ [MeY]2-+ HInd-+2H+
Лабораторная работа №5. КОМПЛЕКСОНОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ Комплексонометрическое определение общей жесткости воды основано на титровании солей кальция и магния, содержащихся в воде и обуславливающих ее общую жесткость, раствором комплексоиона III (трилон Б). В качестве индикатора используется хромоген черный (эриохром черный). Сa2+ + HInd2- → CaInd- + H+ винно-красный Константа нестойкости комплексов металлов с индикаторами: Сa2+ 3, 9*10-6 Mg2+ 1, 0*10-7 Константа нестойкости комплексов Ca2+, Mg2+ с комплексоионом III меньше (2, 7*10-11, 2*10-9). Поэтому при титровании комплекса металлов с индикатором нарушается и образуется более прочные комплексы с комплексоионом III. CaInd-+Na2[H2Y] → Na2[CaY] + Hind- + H+ винно-красн синий В эквивалентной точке винно-красная окраска растворов сменяется синей вследствие накопления анионов индикатора. При реакции образования комплексов металлов с комплексоном III в растворе накапливаются ионы водорода, поэтому титрование следует проводить при рН=8-10 (комплексные соединения металлов с комплексоном III в кислой среде неустойчивы). Титрование следует проводить в присутствии аммонийной буферной смеси.
Выполнение определения Отбирают пипеткой 50 мл. анализируемой смеси, переносят в коническую колбу для титрования ёмкостью 200-250 мл., добавляют тмеренные мензуркой 5 мл аммонийного буферного раствора (NH4OH+NH4Cl) и перемешивают. Прибавляют 20-30 мг (на кончике скальпеля) сухой индикаторной смеси, перемешивают раствор до растворения. Раствор, окрашенный в красный цвет, титруют 0, 1н раствором комплексона III до перехода окраски в сине-голубую. В конце титрования раствор комплексона прибавляют по одной капле. Повторите титрование 3 раза и для вычисления возьмите среднее значение. Занесите результаты вычисления в журнал: V Н2О, мл V трилон Б, мл индикатор 50, 0 V1 эрихром черный Т 50, 0 V2 50, 0 V3 ________ Vсреднее Вычисления: Общая жесткость воды (Ca2+, Mg2+ на 1л) вычисляют по формуле в тысячных долях эквивалентной массы:
Сн- нормальность комплексона III V1-объём раствора комплексона III, затраченный на титрование (среднее значение) V-объём воды, взятый на анализ. (1мл 0, 1н раствора комплексона III соответствует 0, 1 мг/экв солей калия и магния).
КОНТРОЛЬНАЯ ЗАДАЧА Определение содержания кальция и магния при их совместном присутствии в растворе.
Анализируемый раствор представляет смесь растворов двух солей: хлористого кальция (СаСl2) и хлористого магния (МgCl2).Для раздельного определения в этой смеси содержания кальция и магния определяют в одной части раствора содержания кальция, а в другой части определяют содержание суммы кальция и магния. Для этого титруют две аликвотные части анализируемого раствора раствором комплексона III с применением различных металлиндикаторов. 1. Для определения содержания кальция титрование проводят с применением индикатора мурексида. 2. Для определения содержания кальция и магния титрование проводят с применением индикатора эриохром черный Т. Содержание магния вычисляют по разнице между объёмами раствора комплексона III, пошедшего на титрование суммы кальция и магния-V2 и на титрование одного кальция V1. V-число мл раствора комплексона III, эквивалентное содержанию магния V=V2-V1 В качестве титранта применяют раствор комплексона III. Определение содержания кальция в присутствии магния основано на применении индикатора- мурексида. Мурексид не образует окрашенного комплексного соединения с ионами Mg2+. С ионами Са2+ мурексид образует окрашенное в красный цвет комплексное соединение. Кн=7, 9 Константа нестойкости комплексного соединения кальция с комплексоном III (2, 57 CaInd++[H2Y]2- → [CaY]2- + Ind- +2H+ красн безцвет. синие-фиолет. При титровании в растворе накапливаются ионы водорода, поэтому титрование проводится в щелочной среде рН 11-13, т.к. комплексное соединение кальция с комплексоном III неустойчиво в кислой среде.
Выполнение определения Раствор смеси солей кальция и магния в мерной колбе на 100 мл разбавляют водой до метки. а) Определение кальция (Са2+). Аликвотную часть 10мл переносят в коническую колбу, добавляют 40мл дистиллированной воды, 10мл 20% раствора КОН, перемешивают, на конце шпателя вносят 20-30 мг индикатора мурексида. Титруют раствором комплексона III до перехода красно-розовой окраски в фиолетовую. Титрование повторяют три раза, берут среднее число миллилитров (V1). V1.1 V1.2 V1..3 ______ V1cр. Молярная масса эквивалента Са2+ в реакции образования комплекса с комплексоном III равна:
б) Определение содержания магния Мg2+ (титрование суммы кальция и магния). Аликвотную часть 10 мл переносят в коническую колбу, добавляют 40 мл воды, 10 мл аммиачного раствора, перемешивают. Добавляют на кончике шпателя 20-30 мг эриохрома черного Т, перемешивают и титруют 0, 1 н раствором комплексона III до перехода винно-красной окраски в сине-голубую. Титрование повторяют три раза и берут среднее число. По разности между объёмами комплексона III, пошедшими на титрование суммы Са2+ и Мg2+ (V2) и одного кальция Са2+ (V1) находят объём комплексона III, эквивалентный содержанию магния Mg2+. V2.1 V2..2 V2..3 ______ V2cр. Молярная масса эквивалента Mg2+ равна:
Вычисления. ТкомплексонаIII/Са= ТкомплексонаIII/Mg= Содержание Са = Содержание Mg = А- аликвотная часть анализируемого раствора (10 мл), взятая для титрования.
Вопросы к разделу
1. На чем основан метод комплексонометрического титрования? 2. Что такое комплексоны? 3. Какие ионы можно определять комплексонометрическим методом? 4. Что представляют собой металлиндикаторы и на чем основано их действие? 5. В чем отличие металлиндикаторов и рН-индикаторов? 6. Какие требования предъявляются к металлиндикаторам? 7. Что такое общая жесткость воды? В каких единицах она выражается? 8. Присутствием каких солей обусловлена карбонатная жесткость воды? 9. В какой среде проводится определение общей жесткости воды? 10. На чем основано определение момента конца титрования при определении жесткости воды?
ГРАВИМЕТРИЧЕСКИЕ МЕТОДЫ
Введение
Гравиметрия является простым и точным, хотя довольно продолжительным методом анализа. Сущность гравиметрии заключается в том, что определяемую составную часть анализируемого вещества изолируют либо в чистом виде, либо в виде соединения определенного состава, которое затем взвешивают. Гравиметрические методы подразделяют на методы отгонки и осаждения. Наибольшее значение имеют последние. В этих методах определяемый компонент выделяют в осадок в виде малорастворимого соединения, которое после соответствующей обработки (отделение от раствора, промывание, высушивание или прокаливание) взвешивают. При осаждении всегда нужно брать некоторый избыток осадителя. Для получения чистых однородных крупнокристаллических осадков (если вещество кристаллическое), или хорошо скоагулированных осадков (если вещество аморфное) необходимо соблюдать ряд правил. Состав вещества, подлежащего взвешиванию (гравиметрическая форма), должен строго отвечать определенной химической формуле. В гравиметрии осаждение проводят, как правило, из горячих разбавленных растворов медленным разбавлением раствора осадителя при непрерывном перемешивании. Перед осаждением в раствор добавляют вещества, повышающие растворимость осадка (в случае осаждения кристаллических осадков), или способствующие коагуляции коллоидных растворов (в случае осаждения аморфных осадков). В условиях аналитического осаждения кристаллических осадков осадок быстро формируется и поэтому образуются кристаллы разных размеров и несовершенные по форме. Для улучшения структуры осадка применяют старение осадка, т.е. настаивание осадка под маточным раствором. при этом уменьшается общая поверхность осадка за счет укрупнения кристаллов и совершенствуется форма кристаллов. Аморфные осадки склонны к загрязнению, поэтому их отфильтровывают сразу же после осаждения. Результаты гравиметрических определений чаще всего выражают в абсолютных величинах или в процентах к навеске вещества. Например, если в силикате определяют содержание SiO2, то для вычисления пользуются формулой
так как гравиметрической формой является определяемое вещество. Однако часто массу определяемого компоненнта непосредственно не взвешивают. Например для определения сульфат ионов взвешивают осадок сульфата бария. поэтому для пересчета массы осадка в массу определяемого компонента вводят гравиметрический фактор F, который равен
где a и b – целые числа, на которые умножают числитель и знаменатель, чтобы число молей в числителе и знаменателе было химически эквивалентно. Например, если гравиметрической формой является Mg2P2O7, то для пересчета в MgO можно использовать Величину навески пробы для выполнения одного определения можно рассчитать по фомуле
где g-искомая навеска; Масса гравиметрической формы определяется с одной стороны погрешностью весов, с другой – оптимальной массой осаждаемой формы. погрешность обычных аналитических весов составляет 1·10-4 г. Поскольку относительная погрешность определения обычно не превышает 0, 1 %, погрешность весов должна составлять не более 0, 1 % от минимальной массы гравиметрической формы. Отсюда
В зависимости от структуры осадка масса осаждаемой формы может колебаться в следующих интервалах (в г): аморфный (Fe2O3·nH2O и т.п.)………………………….0, 07 – 0, 1 кристаллический, легкий (CaCO3 и т.п.)……………….0, 1 – 0, 15 кристаллический, тяжелый (BaSO4 и т.п.)……………..0, 2 – 0, 4 кристаллический очень тяжелый (PbSO4 и т.п.)……….до 0, 5. Эти примерные критерии служат для оценки массы гравиметрической формы и массы пробы соответственно. Например, какую навеску стали, содержащей около 0, 5 % никеля следует взять для определения никеля в виде диметил глиоксимата? Диметилглиоксимат никеля – мелкокристаллическийосадок внешне напоминающий аморфный, поэтому можно принять массу осаждаемой формы равной 0, 1 г. Гравиметрическая и осаждаемая формы в данном случае совпадают, следовательно m= 0, 1 гравиметрический фактор F=0, 2032. Отсюда величина навески
ТЕХНИКА РАБОТЫ
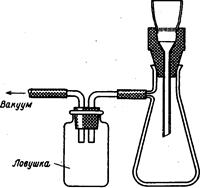
Взятие точной навески. Одним из основных приборов в количественном анализе являются аналитические весы – прибор для определения массы вещества. обычно они позволяют определить массу с точностью до 0, 0001 г. правила обращения с аналитическими весами. 1. Нагрузка на чашки весов не должна превышать предельной, указанной для данной системы весов. 2. Взвешиваемый предмет и разновески можно помещать на чашки весов и снимать с них только при закрыттом арретире. 3. Температура взвешиваемого предмета и температура окружающей среды должны быть одинаковыми (нельзя взвешивать теплые предметы). 4. Взвешиваемые вещества должны находится в чистой, сухой таре (бюксы, тигли, часовые стекла и т. п.). Вещества нельзя непосредственно помещать на чашки весов. Летучие и гигроскопичные вещества нужно взвешивать в хорошо закрытых сосудах. 5. Дверцы весов во время взвешивания должны быть закрыты. 6. Все взвешивания для данного анализа должны проводится на одних и тех же весах. Весы нужно содержать в чистоте, случайно-рассыпаное вещество нужно удалить спкциальной кисточкой. При неисправности весов нужно обращаться к преподавателю. Анализ начинается со взятия точной навески анализируемого вещества. Перед этим необходимо выбирать ее примерную величину, которая зависит от ряда факторов, в частности величина навески определяется примерным содержанием вещества в пробе. В общем случае навеска не должна быть не слишком большой, ни слишком малой. Величина навески зависит от характера осаждаемой формы. Осадок должен выделяться в таких количествах, чтобы его можно было легко отфильтровать и промыть. Поэтому, если речь идет о кристаллических осадках, (типа BaSO4), то нужно брать такую навеску, чтобы масса гравиметрической формы составляла 0, 3-0, 5 грамма. Если выделяется объемный аморфный осадок, например Fe2O3·nH2O, то навеска анализируемого вещества должна быть такой, чтобы масса гравиметрической формы составляла 0, 1-0, 2г. Навеска одного и тогоже анализируемого вещества может быть различной в зависимости от выбранного метода анализа. Если осаждают аллюминий в виде гидроксида (аморфный осадок), навеска должна иметь одну величину, если этот осадок осаждают 8-оксихинолином (кристаллический осадок) – другую. Навески твердых веществ берут обычно в бюксах, стеклянных стаканчиках, на часовых стеклах. Бюксами пользуются обязательно при взвешивании гигроскопичных и летучих веществ. Существуют два основных метода взятия точной навески. 1. Взвешивают на аналитических весах (с точностью до 0, 0001 г) чистый сухой бюкс (стаканчик, часовое стекло). Затем помещают его на технические весы, насыпают на него и отвешивают (с точностью до 0, 01 г) анализируемое вещество. После чего бюкс с веществом взвешивают на аналитических весах (с точностью до 0, 0001 г). Разность двух взвешиваний на аналитических весах дает массу взятой навески. Взвешенное вещество осторожно, не распыляя, пересыпают в химический стакан (или через сухую воронку в колбу), после чего смывают в стакан с водой из промывалки оставшиеся в бюксе и воронке частицы вещества. 2. На технических весах вначале взвешивают пустой бюкс, а затем с анализируемым веществом. Помещают бюкс с веществом на аналитические весы и взвешивают (с точностью до 0, 0001 г). Осторожно пересыпают, не распыляя, вещество в стакан (или через сухую воронку в колбу). Бюкс с оставшимися частицами вещества вновь взвешивают на аналитических весах. По разности двух взвешиваний на аналитических весах находят массу навески. Этот способ называется «взятием навески по разности», он наиболее часто применяется в гравиметрии. Жидкости для анализа тоже берут по разности, всегда пользуясь для этого бюксами. Переведение навески в раствор Важным этапом анализа является выбор растворителя для растворения анализируемого вещества. Некоторые вещества растворимы в воде, но чаще для растворения приходится использовать другие растворители, их нужно выбирать, чтобы растворение было полным. При выборе растворителя нужно учитывать и химический состав анализируемого материала. Например, не рекомендуется применять кислоту, если исследуемый материал содержит мышьяк, ртуть (II), так как при растворении эти элементы могут быть частично потеряны из-за летучести их хлоридов. Наиболее часто для растворения используют кислоты: соляную, серную, азотную, хлорную или их смеси. Реже применяют растворы гидроксидов щелочных металлов. Растворение навески обычно проводят в химических стаканах вместимостью 200 – 500 мл, при этом надо следить, чтобы не потерялась часть вещества при растворении за счет разбрызгивания раствора, если реакция растворения протекает бурно или при растворении выделяются пузырьки газов, которые могут увлечь за собой часть раствора. Поэтому стакан обязательно закрывают часовым стеклом, чтобы его выпуклая часть была обращена внутрь стакана. Растворитель добавляют постепенно небольшими порциями, осторожно приливая его по внутренней стенке стакана. Иногда для ускорения растворения следует нагревать содержимое стакана. Для нагревания используют водяную или песочную баню, можно пользоваться электроплиткой с терморегулятором. Нагревание должно быть постепенным и равномерным. После окончания растворения вещества нужно ополоснуть водой из промывалки часовое стекло и стенки стакана, сливая промывные воды в тот же стакан. Иногда после растворения раствор имеет слишком большой объем, в этом случае его следует упарить. Кипение раствора недопустимо, так как могут быть потери вещества за счет разбрызгивания раствора. Ряд веществ невозможно перевести в растворимое состояние с помощью жидких реагентов. В этих случаях прибегают к сплавлению с помощью плавней. В качестве плавней применяют безводные карбонаты калия и натрия (часто их смесь для понижения температуры плавления), гидросульфат, пиросульфат калия и натрия, едкий натр, гидроксид натрия и др., т. е. Вещества кислотного или основного характера в зависимости от состава исследуемого материала. Если анализируемое вещество предлагается сплавлять, его очень тонко измельчают в ступке; для сплавления применяют фарфоровые, кварцевые, платиновые, железные, никелевые тигли. Размер тигля выбирают так, чтобы он был заполнен массой не более чем наполовину. Берут навеску, взвешивая на аналитических весах пустой тигель и тигель с анализируемым веществом (см. выше). Затем в тигель с навеской постепенно, небольшими порциями, вносят 5-8 кратное количество плавня, каждый раз тщательно перемешивая содержимое тигля маленьким шпателем или стеклянной палочкой; последней порцией плавня ополаскивают шпатель или палочку. Закрывают тигель крышкой, ставят в фарфоровый треугольник и слабо нагревают в течении нескольких минут на небольшом пламени газовой горелки, чтобы не произошло потери вещества за счет воды и газов. Затем проводят сплавление при нужной температуре (300 1200 °С) на газовой горелке или электрической печи. По окончании сплавления тигель медленно охлаждают (до темно-красного каления), берут его тигельными щипцами (с платиновыми наконечниками) и медленно вращают, чтобы плав распределился тонким слоем по стенкам тигля для облегчения отделения плава от тигля, после чего тигель медленно охлаждают, погружая его в чашку с холодной водой (это приводит к растрескиванию плава и облегчает его отделение от стенок тигля). Охлажденный тигель помещают в фарфоровую или платиновую чашку и переводят плав в раствор, обрабатывая его водой или раствором кислоты. После полного отделения плава от тигля (или после полного растворения плава) тигель с помощью стеклянной палочки вынимают из чашки и тщательно ополаскивают водой из промывалки над чашкой или стаканом, в котором будут проводить осаждение. К сплавлению стараются не прибегать, так как оно имеет ряд недостатков и в первую очередь тот, что исследуемый раствор, полученный в результате сплавления, значительно загрязнен посторонними веществами, так как плавни, как сказано выше, берутся в больших количествах. Осаждение. В гравиметрии применяют различные осадители. Это могут быть неорганические реагенты, например соляная или серная кислота (для осаждения ионов серебра или бария), хлорид бария (для осаждения сульфат-ионов), водный раствор аммиака (для осаждения гидроксидов) и т. д. Большое значение имеют органические осадители, обладающие рядом преимуществ перед неорганическими. Наиболее часто применяют 8-оксихинолин, диметилглиоксим, 1-нитрозо-2-нафтол, тетрафенилборат натрия и др. Осаждение проводят в химических стаканах вместимостью 200-400 мл, как правило, из горячих разбавленных растворов путем медленного прибавления раствора реагента осадителя при непрерывном перемешивании раствора. Прежде всего следует рассчитеть нужный объем раствора-осадителя с учетом его небольшого избытка (избыток обычно составляет 10 %) Рассчитывают объем растворителя исходя из взятой навески образца и примерного содержания в нем определяемого компонента. Рассчитанное количество раствора осадителя помещают в бюретку и закрепляют ее в штативе (иногда осадитель добавляют не из бюретки, а из пипетки, но это менее удобно и требует хороших навыков работы). Исследуемый раствор разбавляют до нужного объема, добавляют в него вспомогательные реагенты и нагревают не доводя до кипения (чтобы не произошло потерь из-за разбрызгивания раствора), поместив стакан на асбестовую сетку над газовой горелкой или на электроплитку с терморегулятором. Помещают в стакан стеклянную палочку с резиновым наконечником Ставят стакан под бюретку, чтобы носик бюретки находился внутри стакана, и медленно, по каплям добавляют раствор осадителя при непрерывном перемешивании осаждаемого раствора палочкой (следует избегать касания палочкой стенок стакана). Раствор осадителя должен литься по внутренней стенке стакана, а не капать каплями на середину, во избежание разбрызгивания. После добавления рассчитанного количества осадителя следует убедиться в полноте осаждения. Для этого дают осадку немного осесть на дно стакана и к просветленному раствору добавляют несколько капель осадителя, наблюдая, не появится ли муть в местах падения капель осадителя. Если муть не появляется, то можно считать, что полнота осаждения достигнута. Отфильтровывание и промывание осадков. Кристаллические осадки отфильтровывают после их настаивания под маточным раствором в течение нескольких часов. Не вынимая стеклянную палочку, закрывают стакан часовым стеклом и оставляют. Аморфные осадки отфильтровывают через 5 — 10 мин после осаждения, дав осадку собраться на дне стакана. Отделять осадок от раствора можно с помощью специальных фильтрующих тиглей или фильтровальной бумаги. Фильтрующие тигли бывают двух типов: фарфоровые и стеклянные. Первые имеют неглазурованное пористое дно, причем тигли различаются по номерам в зависимости от размеров пор дна и могут быть применены для отфильтровывания осадков различной дисперсности. В стеклянных тиглях дно представляет собой пористую стеклянную пластинку; они также различаются по номерам в зависимости от размеров пор фильтрующей пластинки. Фарфоровые тигли выдерживают прокаливание выше 500 °С, стеклянные тигли можно лишь высушивать при температуре не выше 150 °С. Перед работой тигли тщательно промывают кислотами (соляной или азотной) для очистки пор, иногда раствором аммиака (для очистки от остатков осадка AgCl). Затем их промывают водопроводной водой и, наконец, несколько раз ополаскивают дистиллированной. Промывание тиглей и Отфильтровывание через них осадков проводят с отсасыванием жидкости под вакуумом. Для этого тигель вставляют в просверленную резиновую пробку (прокладку), которой закрывают колбу для отсасывания жидкости, присоединенную через предохранительную склянку к водоструйному насосу (рис. 2.1). Техника фильтрования через фильтрующие тигли аналогична технике фильтрования через бумажные фильтры; доведение фильтрующих тиглей до постоянной массы аналогично доведению до постоянной массы обычных тиглей.
Рисунок 2.1 Стеклянная палочка с резиновым наконечником Популярное:
|
Последнее изменение этой страницы: 2016-06-05; Просмотров: 2146; Нарушение авторского права страницы
 (моль/л)
(моль/л)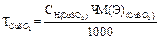 (г/см3)
(г/см3) (г)
(г) (г/см3)
(г/см3)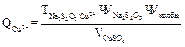 (г).
(г).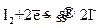 : а) справа налево; б) слеванаправо?
: а) справа налево; б) слеванаправо? 
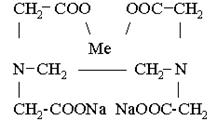
 (мг экв/л)
(мг экв/л)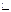 10-6 (рН 10-13)
10-6 (рН 10-13)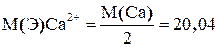 г.
г.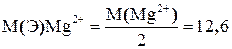 г.
г.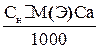 (г/мл)
(г/мл) (г/мл)
(г/мл)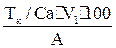 (г)
(г)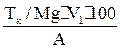 (г)
(г)
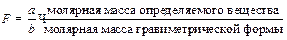
 .
.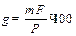 ,
,  , то есть
, то есть 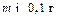 .
.