|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
XI. 1.2. Умственная отсталость при моногенных болезнях
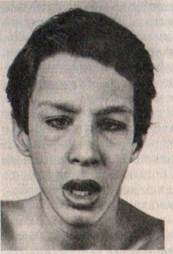
Моногенные заболевания — это гетерогенная группа состояний, различающихся как по специфичности мутаций, особенностям патогенеза, так и по клинической картине. К группе моногенных заболеваний с умственной отсталостью относятся некоторые наследственные заболевания обмена веществ, болезни соединительной ткани, изолированные формы микроцефалии, гидроцефалии и ряд других заболеваний. Как было отмечено выше, среди моногенно наследуемых заболеваний многочисленную группу составляют наследственные дефекты обмена, и в частности энзимопатии или ферментопатии. К началу XXI в. известно более 100 энзимопатий, и более чем для 40 из них принципиально разработаны методы медикаментозной терапииили диетолечения. Ранняя диагностика и своевременно начатое лечение позволяют в большинстве случаев предупредить поражение мозга (и, следовательно, интеллектуальное недоразвитие) на тех этапах его формирования, когда он особенно уязвим. Наследуются энзимопатии чаще всего аутосомно-рецессивно или Х-сцепленно рецессивно, их частота варьирует в широких пределах (от 1: 1000 до 1: 1000000). Центральным патогенетическим звеном энзимопатии является отсутствие или значительное снижение активности того или иного фермента, что блокирует или вызывает существенную недостаточность определенного биохимического процесса. Поскольку большинство ферментных систем многокомпонентны, энзимопатии обычно представлены несколькими генетическими формами. Следует также учесть, что фермент практически всегда вовлечен в несколько метаболических путей, что делает заболевание полисимптоматическим, поражает несколько систем органов. Соответственно этому изолированные нарушения интеллекта встречаются редко; из других систем особенно часто поражается зрение (при галактоземии, гомоцистинурии, мукополисахаридозах, амавротической идиотии и др.). Одной из наиболее часто встречающихся (в среднем 1: 10000) и хорошо изученных энзимопатий является фенилкетонурия (ФКУ, фенилпировиноградная олигофрения, болезнь Фёллинга). ФКУ обусловлена дефицитом фенилаланингидроксилазы (самого апофермента или коферментной системы — НАДФН и дигидробиоптерина). Фенилаланин при этом не может превращаться в тирозин, его содержание в крови повышается (иногда в 35—40 раз), и он включается в побочный метаболический путь (трансаминирование), превращаясь в токсичные для развивающейся нервной системы фенилпиропиноградную, фенилуксусную кислоты и другие фенилкетоновые производные. Главным следствием токсического действия фенилкетокислот является умственная отсталость (в 65 % — глубокая, в 31, 8% — умеренная и тяжелая и только в 3, 2 % — легкая). Фенилпировиноградная кислота выделяется с мочой, придавая ей особый «мышиный» («волчий») запах. Дефицит тирозина сказывается на образовании пигмента меланина, в меньшей степени страдает обмен тиреоидных гормонов и катехоламинов. В связи с этим у больных с ФКУ кожа депигментирована (или слабо пигментирована), обладает повышенной чувствительностью к ультрафиолету, часто развиваются экземы и дерматиты; волосы светлые и светлый цвет радужной оболочки глаза. Череп (особенно его мозговая часть) недоразвит (вторичная микроцефалия). Характерна специфическая поза: локтевые, тазобедренные и коленные суставы слегка согнуты, туловище наклонено вперед. Патологоанатомически обнаруживается малая масса мозга, дефекты миелинизации в коре больших полушарий (особенно в лобных и височных долях) и других структурах (внутренняя капсула, зрительные проводящие пути), депигментация черной субстанции. Психопатологически кроме умственной отсталости отмечается недоразвитие речи (ее или совсем нет, или есть отдельные слова, которые больные не соотносят с объектами), резко нарушено понимание речи и звукопроизношение. Неврологическая симптоматика неспецифична: нередки эпилептиформные припадки, нарушения мышечного тонуса, плахая координация движений, много стереотипии, часты другие знаки экстрапирамидной недостаточности (атетоидные, хореиформные движения). Поведение больных различно. В одних случаях оно близко к полевому (двигательное беспокойство, нецеленаправленные, не- управляемые перемещения от объекта к объекту, бесцельные манипуляции с предметами и т.д.). В других — дети пассивны, вялы, не обнаруживают чувства привязанности, плохо узнают близких, оживляются главным образом при упоминании о еде. В нелеченых случаях первые проявления обнаруживаются чаще всего через 2 — 3 месяца после рождения (редко раньше), и в целом динамика заболевания не укладывается в чисто эволютивные закономерности. Нарушение интеллектуального развития показано и у некоторых гетерозигот — носителей гена ФКУ. При заведомой подтвержденное™ факта гетерозиготного носительства у родителей и сибсов больных в 4 % случаях выявлена легкая интеллектуальная недостаточность, а в 6, 5 % — нижняя границанормы по интеллекту с соответствующим невысоким уровнем образования, профессиональной и социальной адаптации. Начало диетолечения (исключение продуктов, содержащих фенилаланин) в первые недели жизни и проведение его на протяжении 10—12 лет позволяет примерно в 90 % случаях предупредить развитие умственной отсталости; если лечение начинается в старшем возрасте, развитие интеллектуальной несостоятельности предупредить не удается, но поведение несколько нормализуется, реже встречаются эпилептиформные припадки. Успешное применение медикаментозных методов и диетотерапии при ФКУ — один из наиболее ярких примеров медицинской коррекции и профилактики отклонений в развитии. Еще одним примером той же группы расстройств является гомоцистинурия, при которой внешний вид больных напоминает синдром Марфана. Частота гомоцистинурии составляет от 1: 50 000 до 1: 250000. Минимальные диагностические признаки: марфаноидный фенотип, повышение концентрации метионина и гомоцистина в плазме крови в сочетании со снижением того же показателя для цистина, а также повышением экскреции гомоцистина с мочой (гомоцистинурией). В основе патогенеза гомоцистинурии лежит нарушение обмена серосодержащих аминокислот. В норме одна из таких аминокислот — метионин — через ряд промежуточных стадий (в том числе гомоцистеин и цистатионин) переходит в цистеин. В настоящее время известно 4 формы гомоцистинурии, и в большинстве случаев отмечается недостаточность фермента пистатионин-бета-синтазы, в результате чего повышается содержание в крови, тканях и моче гомоцистина (производное гомоцистеина), а иногда и метионина. Повышенная концентрация этих естественных для организма веществ вызывает очаговые некрозы в почках, селезенке, слизистой оболочке желудка, сосудах. Повреждения стенок сосудов, а также активизация свертывающей системы крови усиливают тромбообразование, что проявляется тромбозами коронарных, сонных, почечных артерий, генерализованным венозным тромбозом. Это может приводить к артериальной гипертензии, различным неврологическим нарушениям, ранней смерти. Характерным для гомоцистинурии является также удлинение трубчатых костей, воронкообразная или килевидная деформация грудной клетки, сколиоз, кифоз, вальгусная деформация коленных суставов и/или стоп, плоскостопие, изменение формы и расположения зубов, а также остеопороз, склонность к переломам, ограничение подвижности в суставах. У части больных обнаруживается подвывих хрусталика, иногда сопровождающийся миопией, атрофией зрительного нерва, отслойкой сетчатки и глаукомой. Неврологически выявляются различные знаки очаговой патологии (гемипарезы, гемиплегии, реже судороги), расстройства походки. Эти нарушения часто имеют прогрессирующий характер. Отмечаются также плохая переключаемость, внимания, пониженная работоспособность, некритичность к себе и окружающим. Речь представляет собой ограниченный набор коротких аграмматичных фраз, обычны нарушения звукопроизношения, сниженный словарный запас. У подавляющего большинства больных интеллект снижен (IQ от 70 до 30, т.е. от легкой до выраженной интеллектуальной недостаточности). Сходные, хотя и более полиморфные, клинические проявленияхарактерны для других форм гомоцистинурии, обусловленных нарушением коферментных систем обмена серосодержащих аминокислот. Заболевание наследуется аутосомно-рецессивно. Ген гомоцистинурии локализован в длинном плече хромосомы 21. При некоторых заболеваниях (их называют «болезнями накопления») страдают ферменты катаболизма некоторых веществ, в результате чего последние накапливаются в клетках, нарушая всю их жизнедеятельность. Примером могут служить мукополисахаридозы, болезнь Нимана— Пика и др. Так, при болезни Нимана — Пика (наследуется аутосомно-рецессивно, встречается одинаково часто у мальчиков и девочек) обнаруживается дефицит кислой сфингомиелинидазы и нарушается обмен одного из типов липидов — сфингомиелина. Продукты его неполного распада накапливаются в клетках печени, селезенки, головного мозга, лимфатических узлах, лимфоцитах. Выделяют несколько форм заболевания, различающихся клинически (по времени начала, тяжести висцеральных и невропсихических проявлений), и, по-видимому, неидентичных генетически. Общие для всех симптомы — увеличение печени и селезенки, генерализованное увеличение лимфоузлов. При раннем начале заболевания висцеральные признаки быстро нарастают, умственное и физическое развитие грубо задержано, неврологические расстройства быстро прогрессируют, и больные погибают в возрасте 3 — 5 лет. При юношеской форме помимо висцеральных симптомов наблюдаются знаки поражения нервной системы (судороги, мозжечковые симптомы и др.), но они появляются поздно, развиваются медленно. При висцеральной форме поражение нервной системы не характерно. К моногенным формам олигофрении относят истинную микроцефалию и обтурационную гидроцефалию. Малые размеры мозга отмечаются не менее чем у 2, 5 % детей с интеллектуальной недостаточностью. Причины микроцефалии могут быть различными. 1/10 часть таких случаев не сопряжена с экзогенными поражениями внутриутробного периода и относится к генетически обусловленной «истинной» микроцефалии, которая наследуется аутосомно-рецессивно. У этих больных, как правило, отмечается глубокое общее психическое недоразвитие, а нередко также судорожные припадки, церебральные двигательные расстройства. Действие рецессивного гена истинной микроцефалии проявляется примерно и у 10 % гетерозиготных носителей, его признаки — уменьшенный размер черепа и легкий интеллектуальный дефект. Некоторыми специалистами высказывается мнение, что до 10% всех случаев клинически идентифицированной интеллектуальной недостаточности обусловлено гетерозиготностыо по гену истинной микроцефалии. Примерно 1/з всех случаен врожденных гидроцефалий составляет наследственнаяобтурашюнпая гидроцефалия. Наследование чаще происходит по Х-сцепленному рецессивному типу (т.е. встречается только у мальчиков), однако известны случаи аутосомно-доминантного и аутосомно-рецессивного наследования. Вне зависимости от типа наследования основной патогенетический момент — стеноз сильвиевого водопровода. Отсутствие нормальных условий для оттока цереброспинальной жидкости из первых трех мозговых желудочков обусловливает прогредиентную неврологическую симптоматику, и без хирургического устранения дефекта в раннем возрасте прогноз неблагоприятен. К наследственным заболеваниям с умственной отсталостью относят и синдром Мартина — Белл (синдром ломкой Х-хромосомы). Синдром наследуется Х-сцепленно рецессивно и встречается в основном у мальчиков, хотя выявляется и у 1/3 женщин — носительниц гена. Частота его составляет 1: 1250— 1: 5000 лиц мужского пола. В настоящее время выяснен характер генетических изменений, лежащих в основе этого заболевания. Показано, что клинические проявления синдрома связаны с увеличением числа тринуклеотидных повторов цитозин-гуанин-гуанин на определенном участке длинногоплеча Х-хромосомы (Xq27.3). Фенотипически характерны долихоцефальный череп, длинное лицо, большие оттопыренные ушные раковины, выступающий лоб, крупный нос, толстые губы, массивный подбородок (рис. XI.4), макроорхидизм (увеличенные яички), во многих случаях увеличенные кисти и стопы, иногда гинекомастия и ожирение.
Рис. XI.4. Синдром ломкой Х-хромосомы (синдром Мартина—Белл). Большие ушные раковины, выступающий лоб, длинный нос, массивный подбородок
Неврологически чаще всего отмечаются мышечная гипотония, явления оральной апраксии, гидроцефалия. Во всех случаях отмечается умственная отсталость, однако глубина ее различна. Так, для больных мальчиков с синдромом ломкой Х-хромосомы умственное недоразвитие может варьировать от умеренной до глубокой (IQ от 70 до 35), в то же время у лиц женского пола с этой патологией отмечается только легкая интеллектуальная недостаточность. Частой особенностью речи больных являетсяклаттеринг-синдром, при котором отмечается поспешная неразборчивая речь с дизритмией, персеверациями, трудностями подбора слов, расстановки логических ударений. Нередко отмечается шизофреноподобная симптоматика, а также проявления детского аутизма. Даже в тех случаях, когда в клинической картине ведущим признаком являются интеллектуальные нарушения, некоторые из свойственных аутизму особенностей (сензитивность, высокая пресыщаемость, недостаточные коммуникативные возможности, иногда наличие особых интересов) весьма существенны, и их необходимо учитывать в психолого-педагогической работе. В динамике нередко отмечают тенденцию к снижению интеллектуального уровня, что, однако, требует специального изучения: не исключено, что помимо биологических причин существенны неадекватные методы обучения и воспитания, применяемые на протяжении длительного времени (несколько лет). Пример. В семье первый ребенок — мальчик (на момент обследования в возрасте 18 лет) — страдает синдромом Мартина—Белл. Его интеллект соответствует выраженной умственной отсталости, отмечается аутизм; несмотря па официально признанную необучаемость, в условиях специального учреждения для детей с аутизмом удалось сформировать навыки речи, письма, чтения, бытовые навыки; будучи музыкальным, ребенок овладел элементарными навыками игры на фортепиано. В течение года работал дворником и уборщиком служебных помещений, с работой справлялся удовлетворительно, но требовал постоянного контроля. Уровень его социальной адаптации исключал возможность самостоятельной жизни. Родители обратились за медико-генетической консультацией в связи с желанием иметь второго ребенка. Были определены вероятность рождения больного ребенка и меры контроля течения беременности. Исследования в ходе последовавшейбеременности (плодом мужского пола) дали положительные результаты на наличие синдрома ломкой Х-хромосомы, и беременность была прервана; патоморфологические исследования полностью подтвердили правильность пренатальной диагностики. Третья беременность, которая также протекала под медико-генетическим контролем, завершилась рождением нормальной девочки, и последующий пятилетний катамнез указывает на отсутствие у ребенка существенных отклонений в умственном и физическом развитии. Среди многообразных форм умственной отсталости моногенной природы выделяют так называемые ксеродермические формы, при которых интеллектуальный дефект сочетается с поражением кожи. Примером этих заболеваний являются нейрофиброматоз и туберозный склероз. Для нейрофиброматоза (болезнь Реклингхаузена) характерно наличие множественных опухолей по ходу периферических нервов, опухолей центральной нервной системы, органов зрения, внутренних органов, отмечаются пигментация кожи, кожные невусы, костные аномалии. Развернутая форма нейрофиброматоза встречается с частотой 1: 2500—1: 3000 новорожденных, наследуется аутосомно-доминантно со 100 %-й пенетрантностью и вариабельной экспрессивностью. В настоящее время выделяют две формы нейрофиброматоза: классическую периферическую (нейрофиброматоз 1), ген которой локализован на 17-й хромосоме, и центральную форму (нейрофиброматоз II), ген которой находится на 22-й хромосоме. Минимальные диагностические признаки нейрофиброматоза: наличие на коже более 5 пятен «цвета кофе с молоком» диаметром не менее 15 мм; две и более нейрофибромы; глиома зрительного нерпа. Заболевание проявляется с рождения или в первое десятилетие жизни образованием на коже пигментных пятен, размер и число которых постепенно увеличиваются (рис. XI.5, а). Пятна локализуются чаще всего на закрытых участках кожи (на спине и боковых участках туловища, а также в подмышечных и паховых областях). Кожные и подкожные опухоли располагаются по ходу периферических нервов (рис. XI.5, б). Наиболее характерны глиомы зрительного нерва, возможны нейрофибромы на веках, конъюнктиве, роговице глаз, радужке. Если опухоли возникают внутри орбит, то они могут спровоцировать птоз, паралич глазных мышц. Реже возможны скелетные нарушения (кифоз, сколиоз, нарушения трубчатых костей и др.). Поражения нервной системы разнообразны по спектру, выраженности и динамике, что определяется локализацией и размером новообразований. Одним из проявлений локализации их и центральной нервной системе является снижение интеллекта, нарушение памяти, внимания, иногда судорожный синдром. Эти признаки могут проявляться не у всех больных, начинаются они с незначительных проявлений, но, постепенно нарастая, приводят к расстройствам речи, ослаблению отдельных высших психических функций и, как следствие, к трудностям обучения. С течением времени во многих случаях школьные проблемы нарастают, усугубляются личностными отклонениями и могут приводить к переводу на программы более низкого уровня (и соответственно в специальную (коррекционную) школу VIII вида) и/или индивидуальное обучение.
Рис. XI.5. Нейрофиброматоз Реклинтаузема: а — пигментные пятна на коже ребенка; б — кожные опухоли у взрослого больного
Характерной особенностью нейрофиброматоза IIтипа является образование опухолей черепно-мозговых нервов и спинного мозга. В клинической картине на первом плане различные неврологические расстройства, прогрессирующее снижение интеллекта и распад психики в целом. Кожных опухолей и периферических нейрофибром, как правило, нет. Для специальной педагогики этот тип заболевания существенного значения не имеет. Минимальными диагностическими признаками туберозного склероза (болезнь Бурневилля —Прингла) являются ангиофиброма лица, судороги и умственная отсталость. Частота туберозного склероза при рождении составляет примерно 1: 10000, среди всего населения — от 1: 30 000 до1: 100000. 80 % случаев связаны с мутацией de novo, механизм наследования аутосомно-доминантный со 100%-й пенетрантностью и вариабельной экспрессивностью. Проявляется заболевание ввозрасте 2 — 5 лет чаще всего (93%) судорожными припадками различного типа (большие, малые, валаамовы и др.). Из других неврологических нарушений встречаются гидроцефалия, пирамидные и экстрапирамидные симптомы. В головном мозге — в стенках желудочков, мозжечке, базальных ганглиях обнаруживаются многочисленные кальцификаты. Умственная отсталость обнаруживается примерно в 3/4 случаев и может варьировать от легкой до выраженной степени. Различна и се структура: в одной части случаев преобладают явления недоразвития с сохранением типичной для олигофренического дефекта иерархической структурой, в другой части на первый план выхолят процессуальные явления деменции. Если говорить о поражениях кожи, то это прежде всего (70% случаев) ангиофиброма щек в виде " бабочки" (красные и розовые папулы), шагреневая кожа, депигментированные пятна, кофейные пятна, подкожные фиброматозные узелки и др. (рис. XI.6). В зрительной системе встречаются опухолевидные изменения сетчатки, иногда глаукома. Значительно чаще, чем в среднем в популяции, встречаются как доброкачественные, так и злокачественные опухоли различных органов. Течение заболевания прогрессирующее, большинство больных умирают в 20—25 лет.
Рис. XI.6. Туберозный склероз. Поражение кожи щек в виде " бабочки", шагреневые утолщения кожи, пигментированные и депигментированные пятна неправильной формы
Популярное:
|
Последнее изменение этой страницы: 2016-03-25; Просмотров: 1139; Нарушение авторского права страницы