|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
XI. 1.3. Умственная отсталость при дизморфических синдромах
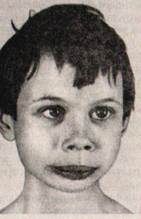
Синдром Прадера—Вилли был выделен в самостоятельное заболевание в 1956 г. Основными клиническими проявлениями синдрома являются мышечная гипотония, гипогонадизм, ожирение, умственная отсталость, уменьшенные размеры кистей и стоп и множественные признаки лис морфогенеза (рис. XI.7). В настоящее время частота синдрома среди новорожденных оценивается примерно 1: 10000 до 1: 20 000; мальчики и девочки болеют одинаково часто. Синдром Прадера — Вилли является классическим примером болезней импринтинга (см. гл. X, разд. «Наследственные болезни с нетрадиционным типом наследования»). Генетически синдром гетерогенен, но большая часть случаев связана с наследованиемвнутрихромосомнои делеции критического региона длинного плеча хромосомы 15(15ql l-ql3) отцовского происхождения. В клинических проявлениях можно выделить две фазы. Первая, ранняя фаза включает антеперинатальный период и первые два года жизни больного и характеризуется слабой подвижностью плода, низким весом при рождении (в среднем составляющим 1900г), опасностью развития асфиксии в результате глубокой гипотонии. В неонатальном периоде отмечаются гипорефлексия, проблемы, связанные с кормлением (дети плохо сосут из-за слабости сосательного и глотательного рефлексов). Дети почти не совершают спонтанных движений, грубо отстают в развитии статических илокомоторных функций. В типичных случаях дети с синдромом Прадера—Вилли начинают держать голову не ранее чем в 6 месяцев, сидеть — после года, ходить — на 3 —4-м году. По достижении 1, 5 — 2 лет проявления мышечной гипотонии смягчаются, развивается неконтролируемая полифагия: больные постоянно испытывают чувство голода, могут есть практически непрерывно. Быстро развивается ожирение, масса тела может превышать норму в 2 — 2, 5 раза. Жир откладывается преимущественно на туловище и проксимальных отделах конечностей. У мальчиков отмечается гипоплазия полового члена и мошонки, крипторхизм (неопущение яичек в мошонку), у девочек — гипоплазия половых губ. У большинства женщин — аменорея, в 50% случаев — гипоплазия матки. Из признаков дисэмбриогенеза у больных наиболее часто регистрируются долихоцефалия, гипертелоризм, эпикант, миндалевидный разрез глазных щелей, «карпий» рот, высокое нёбо, гипопигментация и аномалии дерматоглифики. Рис. XI.7. Синдром Прадера— Вилли. Ожирение, гипогонадизм, маленькие кисти и стопы Почти все больные с синдромом Прадера—Вилли — умственно отсталые, но оценки IQ могут колебаться в широком диапазоне (20 — 90). Часто интеллектуальные трудности представляются более выраженными, чем они есть на самом деле, из-за повышенной пресыщаемости и утомляемости. Если учитывать эти особенности при построении учебного о процесса, то можно добиться значительно большего прогресса в сравнении с исходно ожидаемым. Больные, как правило, добродушны и доброжелательны, безынициативны, часты резкие немотивированные смены настроения. Особенности эмоциональной сферы в ряде случаев приводят к ошибочному диагнозу «аутизм», хотя аутистические черты личности встречаются нередко. Продолжительность жизни больных в среднем составляет 25 — 30 лет. Для взрослых больных на фоне умственной отсталости различной степени тяжести характерны эмоциональная неустойчивость, гиперфагия, сниженная познавательная способность и моторная активность. Синдром Ангельмана выделен в самостоятельное заболевание в 1965 г. Первоначальное его название — синдром «счастливой куклы» из-за характерных клинических проявлений: приступы неконтролируемого смеха, резкие судорожные движения рук, необычная походка, хлопанье п ладоши и специфическая гримаса. Название «синдром Ангельмана» он получил в 1982 г. Его частота составляет 1: 20000 новорожденных. Как и синдром Прадера— Вилли, он представляет собой одну из болезней импринтинга (см. гл. X. разд. «Наследственные болезни с нетрадиционным типом наследования»). Его возникновение у ребенка обусловлено наследованиемвнутрихромосомнои делеции критического региона длинного плеча хромосомы 15(lSqll-ql3) материнского происхождения. Основными клиническими проявлениями синдрома Ангельмана являются задержка умственного и моторного развития, атаксии, гипотония, гиперкинезия, немотивированныйсмех. Наиболее частые признаки дизморфогенеза при синдроме Ангельмана — микробрахицефалия, уплощенный затылок, большая нижняя челюсть, макростомия, частое высовывание языка, редкие зубы и гипопигментация кожных покровов и волос (рис. XI.8).
Рис. XI.8. Синдром Ангельмана у двух братьев. Микробрахицефалия, большая нижняя челюсть, макростомия, мелкие зубы, тонкая верхняя губа, гипопигментация
По мере роста ребенка более заметны становятсянарушения речевого развития, постепенно нарастает тяжесть неврологическойсимптоматики и умственной отсталости, которая достигает в некоторых случаях степени идиотии. Наиболее характерными признаками синдрома Вильямса (синдром «лица эльфа», идиопатической гиперкальциемии) являются необычное лицо, надклапанный стеноз аорты или легочной артерии, повышенное содержание кальция в плазме крови. Популяционная частота 1: 10000, распределение по полу равномерное. Предполагаемый тип наследования аутосомно-доминантный. Для новорожденного характерны малые величины антропометрических показателей (например, масса тела обычно не превышает 2700 г), в дальнейшем развитии всегда отмечается малый рост. Из других особенностей необходимо отметить эпикант, короткий нос с открытыми вперед ноздрями, широкую верхнюю и маленькую нижнюю челюсти, макростомию, полные щеки, оттопыренные уши (рис. XI.9). У всех больных отмечается интеллектуальная недостаточность: чаще всего IQ в пределах 30— 50 (что соответствует имбецильности), но возможны и более легкие (на уровне дебилытости) случаи. Речь больных с синдромом Вильямса имеет довольно большой словарный запас, больные обычно словоохотливы, говорливы, но речевая продукция, по существу, представляет собой более или менее обширный набор речевых штампов, употребляемых часто невпопад, вне связи с ситуацией. Личностно дети с этим синдромом обычно добродушны, приветливы, послушны, у большинства присутствует хороший музыкальный слух. Отмечаются разнообразные неврозоподобные расстройства (страхи, навязчивости, энурез и т.п.). В школьном обучении дети менее успешны, чем это можно было бы предполагать исходя из их личностных особенностей и уровня интеллекта, из-затрудностейорганизации учебного и трудового процессов, повышенной утомляемости и пресыщаемости.
Рис. XI.9. Синдром Вильямса. Эпикант, открытые вперед ноздри, широкая верхняя челюсть, полные щеки, макростомия
Синдром Корнелии де Ланге встречается с частотой 1: 10 000-30000 новорожденных. Генетически синдром, по-видимому, гетерогенен. Большинство случаев спорадические, в ряде случаев обнаруживаются микроструктурные хромосомные перестройки, вовлекающие участок длинного плеча хромосомы 3(3q26.3). Синдром характеризуетсямикроцефалией (чаще в сочетании с брахицефалией), особенностями лица (сросшиеся брови, длинный фильтр, тонкая загнутая внутрь верхняя губа, вывернутые наружу ноздри), гипертрихозом (рис. XI. 10). При рождении масса тела, как правило, в пределах 2100—3200 г, в дальнейшем отмечается отставание в росте. Характерны особенности строения кисти: 1-й палеи расположен проксимально, 2-й палец необычно короткий, мизинец искривлен; иногда отмечается олигодакгилия. Для больных с синдромом Корнелии де Ланге характерна интеллектуальная недостаточность. Примерно в 80 % всех случаев синдрома регистрируется умеренная и тяжелая умственная отсталость IQ (30—50), но есть случаи и с более высокими показателями IQ (75—80). Часто обнаруживается склонность к агрессивным формам поведения (особенно к аутоагрессии), стереотипии, иногда наблюдаются судорожные явления.
Рис. XI. 10. Синдром Корнелии де Ланге. Синофриз, длинные ресницы, маленький нос с открытыми вперед ноздрями, длинный фильтр, большие промежутки между зубами
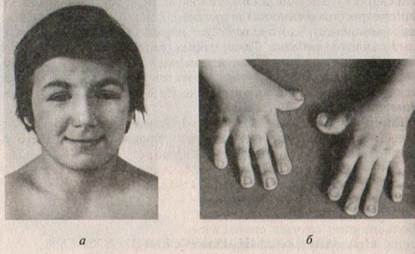
Синдром Рубинштейна—Тейби характеризуется прогрессирующей умственной отсталостью, микроцефалией, широкими концевыми фалангами первых пальцев кистей и стоп, характерным лицом, отставанием в росте (рис. XI. 11). Синдром Рубинштейна—Тейби встречается с частотой 1: 25 000 — 30000 новорожденных. Предположительно синдром наследуется аутосомно-доминантно, ген локализован в коротком плече хромосомы 16. Клинически отмечается микроцефалия, брахицефалия, позднее закрытие большого родничка. Характерными особенностями лица являются выступающий лоб, низкий рост волос, приподнятые дугообразные брови, антимонголоидный разрез глаз, длинные ресницы, умеренная ретрогнатия. Кончик носа загнут книзу, во многих случаях крылья носа гипоплазированы, верхняя губа тонкая. Отмечается своеобразное, напоминающее улыбку выражение лица (см. рис. XI. 11). Иногда встречается агенезия мозолистого тела. В строении скелета выявляются такие отклонения, как лордоз, кифоз, аномалии ребер, грудины. Пороки развития встречаются и во внутренних органах: дефекты перегородок сердца, аплазия (односторонняя) почек, патология мочеточника, мочевого пузыря. Умственная отсталость, как правило, глубокая, но известны случаи пограничного снижения интеллекта IQ (70—80). В некоторых случаях больные склонны к агрессивным реакциям, аутотравматизму, частым аффективным вспышкам. В 25 % случаев встречается судорожный синдром.
Рис. XI.11. Синдром Рубинштейна —Тейби: а — микроцефалия, брахицефалия, приподнятые брони, антимонголоидный разрез глазных щелей, тонкая верхняя губа, выражение лица, напоминающее улыбку; 6 — расширение и укорочение концевых фаланг пальцев кистей
Популярное:
|
Последнее изменение этой страницы: 2016-03-25; Просмотров: 1599; Нарушение авторского права страницы