|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
|
|
Архитектура Аудит Военная наука Иностранные языки Медицина Металлургия Метрология Образование Политология Производство Психология Стандартизация Технологии |
ТЕМА № 7. СЕРТИФИКАЦИЯ. ПОРЯДОК СЕРТИФИКАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ.
ЦЕЛЬ: сформировать знания в области сертификации лекарственных средств. . ЗАДАЧИ ОБУЧЕНИЯ ознакомить студентов с: · современной концепцией фармацевтической системы качества; · основными элементами системы качества
ОСНОВНЫЕ ВОПРОСЫ ТЕМЫ 1. Определение понятия «Лекарственное средство». 2. Подтверждение качества ЛС 3. Порядок предоставления мастер-файла ЛС при регистрации препарата 4. Порядок сертификации ЛС. 5. Система сертификации ЛС. 6. Документация, необходимая при сертификации ЛС РАЗДАТОЧНЫЙ МАТЕРИАЛ Лекарственные средства, используемые в Евросоюзе (ЕС), подлежат контролю качества как со стороны регуляторных органов, так и со стороны фармацевтических компаний. Существуют 3 варианта подтверждения качества используемых АФС, ЛС:
Подробно данные процедуры описаны в руководстве Комитета по лекарственным средствам для человека Европейского медицинского агентства (ЕМА) «Обзор требований к сведениям о АФС, ЛС в разделе регистрационного досье по качеству лекарственного препарата». Предоставление мастер-файла АФС, ЛС при регистрации препарата Мастер-файл АФС, также известный как Европейский мастер-файл лекарственного средства (Drug Master File; DMF), является разделом регистрационного досье препарата Общего технического документа (CTD), в котором приведены сведения об использованной в его производстве АФС. Мастер-файл готовится производителем АФС и разделяется на 2 раздела: раздел производителя препарата и закрытый раздел. Последний раздел, содержащий сведения, составляющие коммерческую тайну, производитель АФС напрямую направляет в регуляторные органы. Такой порядок представления сведений об АФС позволяет защитить права и интеллектуальную собственность производителей. Таким образом, при регистрации каждого препарата из конкретной АФС ее производитель обязан представлять мастер-файл АФС в каждый регуляторный орган. Это создает дополнительные технические затруднения, а также увеличивает риск утечки конфиденциальной информации о АФС. Подтверждение качества путем предоставления мастер-файла для вспомогательных веществ невозможно. Предоставление полных сведений о производстве АФС и методах ее контроля при регистрации препарата При регистрации препарата вместо мастер-файла производитель готовой лекарственной формы с согласия производителя АФС может включить в регистрационное досье полные сведения о АФС, включая данные по ее химическому строению, способу получения, способах контроля и валидации производственных и контрольных методик. Такая процедура может быть применена также в случае, если одна компания производит АФС и/или препарат. Предоставление мастер-файла или полных сведений о АФС — единственные варианты подтверждения качества оригинальных АФС и АФС, не описанных в ЕФ. Сертификация на соответствие требованиям ЕФ В настоящее время сертификация на соответствие требованиям ЕФ является предпочтительным способом подтверждения качества АФС. Этот способ подтверждения качества возможен только для АФС, для которых в ЕФ существуют частные фармакопейные статьи. В настоящее время сертификация проводится как для АФС, так и для вспомогательных веществ. Содержание досье, представляемого при сертификации АФС, совпадает со сведениями мастер-файла. Однако в данном случае мастер-файл направляется в EDQM, а также во множество национальных регуляторных органов. Сертификат на соответствие требованиям ЕФ признается в 37 странах Комиссии ЕФ (27 государств ЕС и ЕЭС (Исландия, Норвегия), а также в других странах мира (Австралия, Тунис, Новая Зеландия), заключивших с EDQM соответствующие соглашения, например Меморандум о взаимопонимании. Следовательно, сертификация АФС экономит время всех участников процесса регистрации препаратов. Процедура сертификации независима от регистрации готовых продуктов и позволяет легализовать АФС сразу во всех странах-членах ЕФ и ряде других государств. Порядок проведения сертификации на соответствие требованиям ЕФ установлен постановлением Частичного соглашения по вопросам здравоохранения Совета Европы AP-CSP(07)1 от 21.02.20073. Требования к досье, предоставляемому на сертификацию, содержатся в рекомендациях EDQM PA/PH/CEP (04)1, 4R «Требования к досье по качеству и микробиологической чистоте фармацевтических субстанций», опубликованных в феврале 2007 г. При оценке досье в процессе сертификации используется документация ЕФ (статьи на конкретную АФС, статьи по общим методам анализа (например, оценка содержания примесей, остаточных растворителей), документы Международной конференции по гармонизации (руководство по оценке стабильности ICH Q1, руководства ЕМА (определение генотоксичных примесей CHMP-SWP, контроль отсутствия приона губчатого энцефалита руководства CPMP/CVMP), документы Европейской комиссии (стандарт GMP). Сертификация проводится в нескольких модификациях: общая процедура сертификации на соответствие требованиям ЕФ для химических АФС, вспомогательных веществ, препаратов растительного происхождения; сертификации продуктов с риском губчатого энцефалита (АФС и сырье животного происхождения, реагенты); комбинированная процедура (сертификация на соответствие ЕФ и на отсутствие риска губчатого энцефалита —для АФС, описанных в ЕФ). Все варианты сертификации возможны для производств в любой точке мира. Сертификация неприменима в отношении биотехнологических препаратов, препаратов из тканей человека и готовых лекарственных форм. Структура системы сертификации Головной организацией, осуществляющей сертификацию субстанций на соответствие требованиям ЕФ, является EDQM. Под его эгидой работают следующие элементы системы сертификации:
Наблюдательный комитет В работе Наблюдательного комитета участвуют председатели экспертных групп по вопросам качества, биологическим препаратам, препаратам растительного происхождения, инспекторат GMP/GDP Комитета по лекарственным средствам для человека ЕМА, председатель комиссии ЕФ, руководитель EDQM, приглашенные эксперты, председатели рабочих групп по технической поддержке. В задачи Наблюдательного комитета входят разработки порядка сертификации, назначение экспертов, назначение специалистов в рабочие группы технической поддержки, разработка и пересмотр технических регламентов рабочих групп, вопросы координации деятельности различных элементов системы сертификации. Рабочие группы технической поддержки Группы технической поддержки включают в себя экспертов, имеющих большой опыт в вопросах сертификации. В их задачи входят принятие решений по техническим вопросам сертификации, арбитраж сторон при противоречивых решениях о возможности выдачи сертификата, подготовка технических руководств, определе ние проблем в работе систем -как технических, так и научных и представление их на рассмотрение Наблюдательного комитета. Штат экспертов
ПОРЯДОК СЕРТИФИКАЦИИ Требования к досье Для проведения сертификации должны быть представлены следующие документы:
С 1 января 2008 г. допускается представление досье в электронном виде. Файл должен сопровождаться бумажной версией, если его объем превышает 20 страниц. Также возможно представление соответствующих разделов CTD на препарат в формате Acrobat (PDF). В мае 2007 г. введены дополнения в порядок сертификации производителей, закупающих полуфабрикаты АФС и проводящих ее очистку. В этом случае сведения о производителях сырья также заносятся в сертификат. Соответствие предприятий, выпускающих сырье и полупродукты для поставок в ЕС, требованиям GMP обязательно. АФС гепаринов (включая низкомолекулярные) должны соответствовать требованиям к контролю недавно обнаруженных примесей. Выпускающий контроль качества этих АФС должен включать в себя данные аналитические методики после должной валидации. Данные по выпускающему контролю предоставляются производителям препаратов для последующего представления в регуляторные органы. Предоставление образцов АФС Предоставление образцов на этапе подачи досье в настоящее время не требуется. Однако необходимы гарантии готовности представить образцы по просьбе директората. Представление досье Заявители представляют досье в EDQM. За 1 месяц до подачи досье на сертификацию EDQM по просьбе заявителя может провести консультацию на коммерческой основе (стоимость 1000 евро за одну консультацию длительностью примерно 1 час). Также сотрудник EDQM может проконсультировать заявителя за пределами директората в ходе научных конференций (стоимость 500 евро за получасовую консультацию). Полнота досье проверяется в течение 8 дней после поступления. Если досье неполное, заявителю направляется запрос о дополнительной информации, на который он должен ответить в течение 6 месяцев. На время предоставления недостающих данных срок рассмотрения приостанавливается. В случае отсутствия ответа досье снимается с рассмотрения.Рассмотрение досье по существу начинается сразу после его доукомплектования. Оценка досье производится двумя экспертами, назначаемыми Наблюдательным комитетом. Заявитель должен быть уведомлен о результате рассмотрения досье в течение 5 месяцев. В случае возникновения вопросов по досье заявителю направляется запрос с максимальным сроком на подготовку ответа, равным 6 месяцам. Последующие запросы EDQM должны быть рассмотрены заявителем в срок до 3-х месяцев. Дополнительные материалы, представленные заявителем, рассматриваются в срок до 4-х месяцев. Количественные показатели сертификации:
Сертификат соответствия требованиям ЕФ. В случае положительного решения о возможности сертификации производителю или эксклюзивному дистрибьютору выдается сертификат. Сертификат подтверждает возможность контроля качества конкурентной АФС с помощью соответствующей монографии ЕФ и иногда дополнительных методов анализа. Сертификат не подтверждает соответствие качества. АФС ЕФ и не является документом, подтверждающим соответствие производственной площадки требованиям GMP. Сведения о производственной площадке включаются в сертификат соответствия. Эксклюзивный дистрибьютор АФС может также быть владельцем сертификата при условии, что производитель предоставил ему сведения мастер-файла или CTD и подтвердил установленным порядком свое согласие на эксклюзивную дистрибуцию. Сертификат содержит спецификацию продукта, данные об условиях хранения и упаковке. В сертификате могут быть указаны дополнительные методы анализа по определению примесей, остаточных растворителей, не описанные в соответствующей монографии ЕФ, по которым производители препаратов также должны проводить входной контроль АФС. Кроме того, отдельно приводится перечень тех методов контроля из монографии ЕФ, проведение которых не требуется. Для стерильных материалов указывается в сертификате метод стерилизации и упаковки. Дополнительно указывается, что методы стерилизации были валидированы. Важно отметить, что информация по стерилизации и ее валидации производителем препарата в регистрационном досье предоставляется отдельно. Сертификаты по риску губчатого энцефалита содержат сведения о стране происхождения животных, использованных в производстве АФС, видов животных и их тканей, а также краткое описание производственного процесса (например, для желатинов). Информация по сертификатам (включая отозванные и приостановленные) доступна на сайте EDQM. КОНТРОЛЬ ПО ТЕСТАМ Вариант 1 1. Вещество или смесь веществ, обладающие определенной фармакологической активностью, предназначенные для производства и изготовления лекарственных препаратов, это (2) А. лекарственная субстанция B. вспомогательные вещества C. БАВ D.БАД E.Активный фармацевтический ингредиент 2. Препараты, содержащие биологические белки, а также биосимиляры, называются: (1) А. лекарственные препараты биологического происхождения В. лекарственные препараты органического происхождения С. лекарственные препараты растительного происхождения D. лекарственные препараты природного происхождения Е. банк клеток 3. Химические, фармацевтические и биологические данные включают информацию о: (3) A. системе обеспечения качества на производстве B. клинических испытаниях C. оформление готового лекарственного средства D. описание состава E. стабильности 4. Комплекс требований, гарантирующих идентичность и стабильность производства, контроля качества ЛС с целью обеспечения их безопасности, эффективности, качества и соответствия требованиям регистрационного досье, это: (1) А. Надлежащая производственная практика В. Надлежащая дистрибьюторская практика С. ГФ РК D. Надлежащая инженерная практика E. Надлежащая фармакопейная практика 5. Комплект документов и материалов установленного содержания, представляемый вместе с заявлением на государственную регистрацию ЛС: (1) А. регистрационное досье B. отчет клинических испытаний C. отчет доклинических испытаний D. “drugmasterfile” E.комплект административной информаций Вариант 2 1. Лекарственное средство в определенной лекарственной форме: (1) А. лекарственный препарат В. лекарственная субстанция С.активный фармацевтический ингредиент D.вспомогательное вещество E.балк-продукт 2. К заявлению о государственной регистрации, перерегистрации ЛС прилагаются: (3) А. регистрационное досье в двух экземплярах, содержащее документы и материалы В. образцы ЛС в количествах, необходимых для проведения трехкратного анализа в соответствии с требованиями нормативно-технического документа по контролю за качеством и безопасностью ЛС С.стандартные образцы лекарственных веществ и посторонних примесей, расходные материалы, применяемые при проведении исследований ЛС (в исключительных случаях и на условиях возврата) D. технологическая схема производства E. данные маркетингового анализа 3. Основания для отказа в государственной регистрации: (1) А. более низкие показатели качества и безопасности ЛС, чем регламентированные нормы в ГФ РК, EUR. PH и USP B. насыщенность рынка препаратами данной группы C. высокие цены стоимости ЛС D. низкие цены стоимости ЛС E. занятость регуляторных органов 4. В регистрационное досье (Common Technical Document) входят: (4) A. административная информация (Модуль 1) B. общее содержание, вводная часть, общее резюме по качеству, доклинический обзор, резюме по доклиническим исследованиям и резюме по клиническим испытаниям (Модуль 2) C. качество (Модуль 3) D. отчеты о доклинических исследованиях (Модуль 4) E. заключение 5. Активный фармацевтический ингредиент (API): A. готовое лекарственное средство B. вспомогательное вещество C. лекарственное средство в определенной лекарственной форме D. лекарственная форма E. лекарственное средство, предназначенное для производства или изготовления лекарственных препаратов Вариант 3 1. Примесь (impurity) – это любой компонент субстанции, который не определен химически как субстанция, может представлять собой: (2) A. стабилизирующую субстанцию B. не идентифицированную примесь C. вспомогательную субстанцию D. идентифицированную примесь E. консервант 2. Химические, фармацевтические и биологические данные включают информацию о: (4) A. разработке B. производственном процессе C. доклинических испытаниях D. характеристиках и свойствах E. процедуре и требованиях контроля качества 1. В современную концепцию обеспечения качества лекарственных средств входят следующие стандарты: (3) A. стандарт надлежащего (качественного) производства лекарственных средств B. стандарт надлежащей (качественной) клинической практики C. стандарт надлежащей (качественной) маркетинговой практики D. стандарт надлежаще (качественной) лабораторной практики E. все ответы неверны 4. Упаковка лекарственного средства (ЛС): (4) A. система доставки ЛС B. средство, обеспечивающее процесс обращения ЛС C. комплекс средств, обеспечивающих процесс обращения ЛС D. защищает ЛС от повреждений и потерь E. предохраняет ЛС от воздействия факторов окружающей среды 5. Фармацевтические факторы, влияющие на биологическую доступность лекарственных веществ: (3) A. вспомогательные вещества B. вид лекарственной формы и пути введения C. технология производства D. материальные потери производства E. возраст персонала Вариант 4 1. Проведение оценки и контроля ЛС в ЕС осуществляется: (1) A. USP B. EUR. PH C. FDA D. Европейским агентством по оценке лекарственных препаратов (EMEA) E. ГФ РК 2. Два основных блока информации в разделе химические, фармацевтические и биологические данные (2): A. об активном(-ых) веществе(-ах) B. об эксципиентах C. о маркировке D. о готовом лекарственном средстве E. об упаковке 3. Активный фармацевтический ингредиент (API): (1) A. лекарственное средство в определенной лекарственной форме B. вспомогательное вещество C. лекарственное средство, предназначенное для производства или изготовления лекарственных препаратов D. лекарственная форма E. готовое лекарственное средство 4. Валидация (validation) – документальное оформление: (1) A. действия, дающего высокую степень уверенности в том, что конкретный процесс, метод или система будут постоянно приводить в результатам, соответствующим заранее установленным критериям приемлемости B. отчета о проделанной работе C. плана работы по проведению исследований D. определения срока хранения E. установления условий хранения 5. Выражение “принятая терминология” используется при описании компонентов лекарственных средств: (4) A. для веществ, которые приводятся в Европейской фармакопее, или согласно ГФ РК B. для других веществ - МНН, рекомендуемое Всемирной организацией здравоохранения C. для веществ, которые не имеют МНН необходимо представить данные о том, каким образом и из чего они произведены D. для красителей их обозначение - соответствующим кодом “Е”, присвоенным им Европейской директивой E. для вспомогательных веществ – их химические формулы
Тест 1 вариант 1.А, Е 2.А 3. С, D, Е 4.A 5.A 2 вариант 1.А 2.A, B, C 3. A 4.A, B, C, D 5.E 3 вариант 1.B, D 2.A, B, D, E 3. A, B, D 4.B, C, D, E 5.A, B, C 4 вариант 1.D 2.A, D 3. C 4.A 5.A, B, C, D ТЕМА № 8. Фармацевтическая система качества (ICHQ 10 ). ЦЕЛЬ: сформировать знания в области фармацевтической системы качества. ЗАДАЧИ ОБУЧЕНИЯ ознакомить студентов с: · современной концепцией фармацевтической системы качества; · основными элементами системы качества
ОСНОВНЫЕ ВОПРОСЫ ТЕМЫ 1. Модель фармацевтической системы качества согласно ICH Q10. 2. Основные элементы системы качества 3. Отличие GMP и ISO 9001 4. Система управления изменениями 5. Система корректирующих и предупреждающих действий (САРА-система). 6. Система мониторинга процессов и качества продукции РАЗДАТОЧНЫЙ МАТЕРИАЛ Элементы системы качества. Современная концепция фармацевтической системы качества основана на подходе ICH[1], который заключается в том, что качество зарождается и подтверждается при фармацевтической разработке и оценке эквивалентности, обеспечивается на этапе переноса технологии и при промышленном производстве, оценивается и совершенствуется на протяжении всего жизненного цикла продукта. Данная концепция поддерживается тремя руководствами — ICH Q8 «Фармацевтическая разработка», ICH Q9 «Управление рисками по качеству» и ICH Q10 «Фармацевтическая система качества». В Европейском Союзе, руководство ICH Q10 в 2008 году включено в структуру первого раздела GMP «Управление качеством» а руководство ICH Q9 введено в качестве двадцатого приложения GMP.
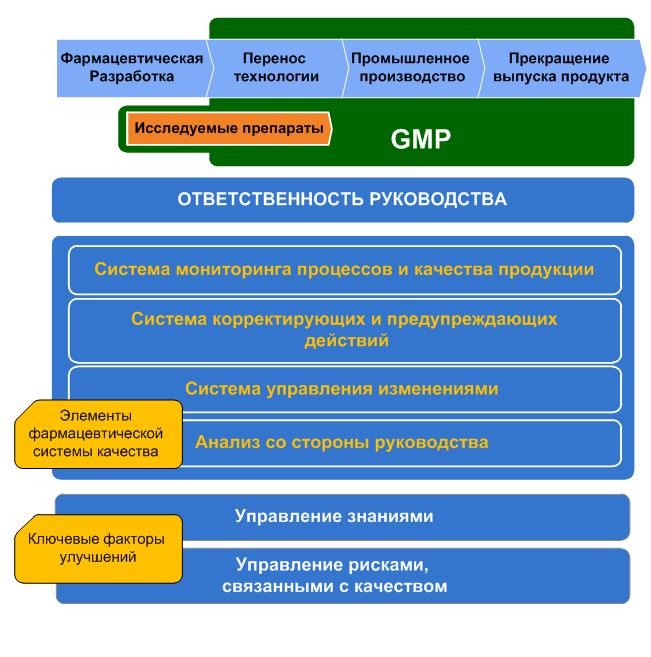
Рис. 1 – Модель фармацевтической системы качества согласно ICH Q10 До 2008 года в Руководстве GMP не было четкой методологии к внедрению системы качества у производителей ЛС. Включение в структуру GMP положений ICH Q10 вводит понятие модель фармацевтической системы качества (см. рис.1), которая может применяться на всех стадиях жизненного цикла лекарственного средства. ICH Q10 вместо«система обеспечения качества»вводит новое понятие «фармацевтическая система качества», которая основана на видении ICH, подходах ISO 9001 и принципах GMP, должна быть четкой и понятной, а также делает акцент на четырех специфических элементах системы (ранее недостаточно описанных в GMP): 1) Анализ со стороны высшего руководства; 2) Система мониторинга процессов и качества продукции; 3) Система управления изменениями; 4) Система корректирующих и предупреждающих действий (САРА-система).
Рис. 2 – Интерпретация модели фармацевтической системы качества, предложенная группой компаний «ВИАЛЕК» В соответствии с ICH Q10 фармацевтическую систему качества необходимо документально оформить в виде Руководства по качеству, принять Политику в сфере качества и подтверждать ее достижением запланированных Целей по качеству. Детальный анализ ICH Q10 показывает, что в целом он содержит элементы, заимствованные из ISO 9001. Де факто ICH Q10 является легализацией ISO 9001 для фармацевтической отрасли (см. таблицу).
Q10 является легализацией ISO 9001 для фармацевтической отрасли В ICH Q10 отсутствует только понятие представителя руководства по качеству (5.5.2 ISO 9001). Очевидно, это связано с описанием деятельности ключевых лиц в Главе 2 GMP, и Уполномоченного Лица в Приложении 16 GMP ЕС, а также необходимостью более тщательных консультаций по данному вопросу. Возможность четкого распределения обязанностей, исключение зон безответственности, стандартизацию различных действий, минимизацию времени на стандартные работы, понимание персоналом своей роли в компании и достижении ее целей и многое другое. Возьмем, к примеру, процесс фармацевтической разработки – четкая стандартизация этапов, структуры документов и ответственности задействованного персонала различных подразделений, значительно сокращают время самой разработки, время на масштабирование, перенос технологии и валидацию процессов, уменьшить количество вносимых изменений – т.е. компании предоставляется возможность оперативно расширять линейку препаратов, заменять один препарат на другой и т.п.В ICH Q10 отсутствует только понятие представителя руководства по качеству (5.5.2 ISO 9001). Очевидно, это связано с описанием деятельности ключевых лиц в Главе 2 GMP, и Уполномоченного Лица в Приложении 16 GMP ЕС, а также необходимостью более тщательных консультаций по данному вопросу. Отличие GMP и ISO 9001, при сравнении которых некоторые специалисты отмечают, что основная идея ISO 9001 заключается в постоянном совершенствовании качества, а задачей GMP является достижение заданного уровня качества, подтверждение его валидацией и поддержание на таком уровне в дальнейшем (т.н. обеспечение воспроизводимости) – т.е. практически, работа по принципу «все лучшее враг хорошего». На самом деле никаких различий нет, и под непрерывным совершенствованием понимается сужение границ фактического разброса значений, выявление источников вариации и их уменьшение, что в результате обеспечивает большую надежность соответствия препарата. Каждая точка, вышедшая из-под контроля – это возможность совершенствования. На такой методологии основана концепция «шести сигм» (Six Sigma), где ставится задача сузить естественную вариацию процесса настолько, чтобы она укладывалась в половину поля допуска. На той же методологии основаны требования к наличию у производителей ЛС спецификации на выпуск, отличной от АНД. Анализ со стороны руководства. Проведение анализа со стороны руководства необходимо для оценки пригодности и эффективности системы качества, а также для выявления и принятия решений об изменениях и улучшениях. С позиции ISO 9001, из которого заимствован сам анализ со стороны руководства, для него приемлемы разные подходы с учетом конкретных особенностей и обстоятельств в компании, поэтому необходима определенная гибкость. Иными словами, анализ следует организовать таким образом, чтобы представителям высшего руководства было удобно его проводить, и чтобы все они в полной мере в нем участвовали. Анализ может быть постоянным процессом и частью повседневной деятельности, или же рассматриваться как редкое событие, либо быть чем-то средним. В ходе анализа со стороны руководства необходимо оценивать: - результаты самоинспекций, внешних надзорных аудитов, и аудитов клиентов; Частью анализа со стороны руководства считается оценка Периодического Обзора качества лекарственного препарата, заявленного в Главе 1 Руководства GMP ЕС. Результатом анализа со стороны руководства, как правило являются решения в виде приказов, распоряжений и планов по улучшению процессов, продукции, распространению необходимых знаний и выделении / оптимизации ресурсов. Система мониторинга процессов и качества продукции. Главным требованием к мониторингу процесса и качества продукции является необходимость проведения всех проверок, испытаний и верификации, а также контроль доступности всей необходимой документации. Целью является возможность продемонстрировать выполнение заданных требований. До сегодняшнего дня, во многих отечественных фармацевтических компаниях преобладает так называемая «инженерная концепция» — контроль соответствия допускам, заявленным в АНД (ФСП). В итоге показатели качества препарата могут сколь угодно варьировать внутри установленных допуском пределов – все, что внутри, считается «достаточно хорошим». Современное же понимание качества предполагает обязательную устойчивость процесса, когда показатели качества препарата получаются близкими, насколько это возможно, к номиналу. В результате, статистически управляемый процесс, будет ориентирован не на границы допуска, а на номинальное значение показателя (центр поля допуска), и всегда находиться в пределах заданных границ, включая те условия, которые создают высокий риск для ошибок производственного процесса. Очень кратко, методология создания системы мониторинга процессов и качества ЛС заключается в следующем: 1) Изучение существующих знаний и информации по препарату и технологическому процессу, самооценка достаточности знаний и их понимания; 2) Выбор стратегии контроля качества; 3) Подбор методов измерений, по возможности их автоматизация; 4) Регламентация плана сбора данных, и правил применения статистических инструментов; 5) Подготовка контрольных листков, сбор и накопление количественных и альтернативных данных по продукту и процессу; 6) Обработка и интерпретация данных; 7) Оценка соответствия и анализ обратной связи. Мониторинг качества продукта подразумевает проведение постоянного контроля на соответствующих этапах процесса производства, контроля, выпуска и хранения для подтверждения соответствия всем установленным требованиям. При этом, чем меньше доверия вызывают результаты прямого контроля продукции, тем более надежным должен быть мониторинг процессов, влияющих на качество такой продукции. Поддержание системы мониторинга процессов и качества ЛС позволит отказаться от такого понятия как плановая ревалидация процесса и плановая реквалификация оборудования. Такой подход основан на принципе, что если по результатам мониторинга объект контроля демонстрирует стабильность, это является подтверждением его воспроизводимости. Ведь данные, накопленные при мониторинге, куда более достоверны, нежели единичные данные одной-двух (ре)валидационных серий. Также, накопленные данные позволяют обосновать пределы спецификаций контроля качества, улучшить понимание процесса и параметров качества препарата, и в последствии отказаться от некоторых точек контроля и поддержать выпуск серии в реальном времени вместо проведения испытаний приемочного контроля (РАТ). Система корректирующих и предупреждающих действий (САРА-система). В компании должна существовать эффективная САРА-система, направленная на устранение причин выявленных и предполагаемых несоответствий. При этом, усилия по поиску необходимых решений и степень документирования должны быть соизмеримы с уровнем риска согласно ICH Q9. Поиск решений должен быть структурированным и направлен на выявление основной причины возникновения несоответствий. Система управления изменениями. Инновации, постоянное совершенствование, данные мониторинга процессов и качества продукта, а также корректирующие и предупреждающие действия всегда ведут к изменениям. ICH Q10 вводит понятие системы управления изменениями, которая позволит проводить необходимую оценку, одобрение и внедрение изменений. Такая система обеспечит своевременность и эффективность непрерывного совершенствования, и в то же время, даст высокую степень уверенности в отсутствии незапланированных последствий любых изменений. Общепринятый подход к внедрению изменений, как правило состоит из 7 шагов: 1) формализация изменения; 2) оценка группой экспертов / оценка рисков, связанных с изменением 3) одобрение / одобрение службой качества; 4) разработка плана внедрения изменения; 5) реализация плана внедрения; 6) верификация / валидация по критериям оценки; 7) документальное закрытие изменения.
ЛИТЕРАТУРА 1. Кодекс «О здоровье народа и системе здравоохранения» 2. Береговых В.В., Мешковский А.П.. Нормирование фармацевтического производства. М.: ИИА Ремедиум, 2001. -527 с. 3. Промышленная технология лекарств в 2-х томах под ред. Проф. В.И. Чуешова. - Харьков: Издательство НФАУ. МТК-Книга. 2002. 4. И.Г. Левашова, А.Н. Мурашко, Ю.В. Пожпружников. Надлежащие практики в фармации. - Киев: Морион. 2006. - 253 с. 5. Shayne cox Gad. Pharmaceutical manufacturing handbook.RegulationsandQuality.Производстволекарственныхсредств.Контроль качества и регулирование. Willey-interscience publication. 2013. - 959 с. 6. Государственная Фармакопея РК УТВЕРЖДЕНА приказом Министра здравоохранения Республики Казахстан от 11 марта 2008 года № 131, Т. 1. - Алматы: Издательский дом «Жибек жолы», 2008. - 592 с 7. «Вызовы и возможности документа ICH Q10фармацевтическая система качества», Александр В. Александров КОНТРОЛЬ ПО ВОПРОСАМ Вариант 1 1. Дать определение понятию «Фармацевтическая система качества». 2. Методология создания системы мониторинга процессов и качества ЛС.
Вариант 2 1. Дать определение понятию «Самоинспекция». 2. Основы современной концепции фармацевтической системы качества.
Вариант 3 1. Дать определение понятию «Ключевые факторы улучшений». 2. Что включает в себя ICH Q8 «Фармацевтическая разработка» ICH Q9 «Управление рисками по качеству»
Вариант 4 1. Дать определение понятию «Фармацевтическая система качества». 2. Что включает в себя ICH Q9 «Управление рисками по качеству» . Вариант 5 1. Дать определение понятию «Фармацевтическая система качества». 2. Система управления изменениями Вариант 6 1. Дать определение понятию «Спецификация качества». 2. Система корректирующих и предупреждающих действий (САРА-система)
Популярное:
|
Последнее изменение этой страницы: 2016-06-05; Просмотров: 1812; Нарушение авторского права страницы